Acido
Enciclopedia on line
In termini generali sostanza dotata di sapore acre (come quello dell’aceto, del succo di limone ecc.), capace di attaccare i metalli (e alcuni loro ossidi) e in grado di reagire con altre sostanze, dette basi, dando luogo a sali. In particolare gli a. inorganici minerali sono formati da idrogeno legato a un non metallo ( idracidi; per es. acido cloridrico), o dall’idrogeno legato a un gruppo atomico, in grado di dar luogo a un anione, che nei casi più comuni ( ossoacidi o ossiacidi; per es. acido solforico) comprende uno o più atomi di ossigeno (per gli acidi organici ➔ carbossilici, acidi).
1. Sistemi acido-base
L’osservazione che l’acidità fosse legata in alcuni composti alla sostituibilità dell’idrogeno da parte dei metalli acquistò un’interpretazione unitaria e quantitativa solo dopo la comparsa della teoria della dissociazione elettrolitica di S.A. Arrhenius (1887). Il chimico svedese stabilì che un a. è un elettrolita che in acqua dà luogo al catione H+, una base all’anione OH−. Così la reazione
NaOH + HCl → NaCl + H2O
è esprimibile in termini di ioni da
Na+ + OH− + H+ + Cl− → Na+ + Cl− + H2O.
Dall’equazione ionica si può osservare che una reazione acido-base secondo Arrhenius è riducibile alla combinazione degli ioni H+ e OH−. Così tutte le reazioni acido-base possono essere interpretate per mezzo di un’unica equazione di chimica. L’acqua pura presenta essa stessa una piccola dissociazione che, secondo la teoria di Arrhenius, può essere espressa dall’equazione
H2O ⇄ H+ + OH−;
in condizioni di neutralità le concentrazioni degli ioni idrogeno e di quelli ossidrilici sono uguali; così è possibile affermare che una soluzione è acida, neutra o basica a seconda che la concentrazione degli ioni idrogeno sia maggiore, uguale o minore di quelli ossidrilici. La teoria di Arrhenius presenta notevoli limitazioni a causa del vincolo costituito dal solvente acqua: in effetti si possono avere casi di sostanze non contenenti ioni ossidrile che si comportano da basi in soluzioni non acquose oppure anche casi di acidi forti disciolti in idrocarburi che non danno luogo a ioni idrogeno. Inoltre si è potuto dimostrare che lo ione idrogeno (cioè il protone) è sempre solvatato; nella ipotesi più semplice (idratazione con una sola molecola d’acqua) si forma la specie H3O+, detta ione idronio. Perciò la teoria di Arrhenius può essere applicata solo per reazioni acido-base nel solvente acqua e nel caso che si possa trascurare l’effetto della solvatazione (➔). Nel 1923, il chimico danese J.N. Brönsted sviluppò una teoria secondo la quale un a. è una sostanza in grado di fornire un protone (mentre una base è una sostanza in grado di accettare un protone). Questa teoria svincola il concetto di a. dal solvente esaminato e correla strettamente il comportamento acido a quello basico: infatti un a. è tale solo quando si trova in presenza di una sostanza alla quale può fornire uno o più protoni. Secondo la teoria di Brönsted la dissociazione di un a. in acqua comporta una reazione acido-base tra questo e il solvente, per es.
HCl + H2O → H3O+ + Cl−
in tal modo la reazione di solvatazione può essere considerata all’interno delle reazioni acido-base. I prodotti di una reazione acido-base di Brönsted sono essi stessi acidi e basi, i quali sono tanto più in grado di scambiarsi il protone quanto più è piccola la costante di equilibrio; per tale motivo si parla di coppie coniugate acido-base. Per es., nell’equazione
NH3 + H2O ⇄ NH4+ + OH−
le due coppie coniugate acido-base sono NH4+/NH3 e H2O/OH−. La teoria di Brönsted modifica il concetto di base di Arrhenius e amplia quello di a.; oltre alle specie molecolari risultano acide secondo Brönsted infatti anche specie ioniche, come HS−, HSO4− ecc., in grado di scambiare il protone con una base. Inoltre, per mezzo della teoria di Brönsted si può facilmente interpretare il comportamento anfotero di alcune sostanze come l’acqua e l’ammoniaca che sono in grado, in certe circostanze, di cedere o di accettare protoni; così la reazione di dissociazione dell’acqua è spiegata dall’equilibrio:
2H2O ⇄ H3O+ + OH−;
analogamente, per l’ammoniaca si ha
2NH3 ⇄ NH4+ + NH2−.
Nello stesso 1923 G.N. Lewis ha proposto una definizione di a. capace di comprendere sostanze (per es., il trifluoruro di boro, BF3, e il triossido di zolfo, SO3) che hanno comportamento acido pur non essendo in grado di scambiare protoni. Secondo gli studi effettuati da Lewis un a. è una sostanza la cui molecola è in grado di completare la struttura elettronica esterna accettando una coppia di elettroni (mentre una base è una specie in grado di donare una coppia di elettroni). La definizione di Lewis modifica e amplia il concetto di a.: infatti, in questo contesto, a. non è HCl quanto lo stesso ione H+, che è la specie in grado di accettare un doppietto elettronico; inoltre risultano acide sostanze come BF3 che presentano lacune elettroniche nell’atomo centrale. In pratica, molte reazioni di complessazione possono essere rubricate come reazioni acido-base di Lewis. La teoria di Lewis amplia anche il concetto di comportamento anfotero: sostanze acide come HCl, in certe circostanze sperimentali, possono comportarsi da basi di Lewis per la capacità di un loro atomo (in questo caso il cloro) di mettere a disposizione coppie di elettroni di valenza non legati. La teoria di Lewis è anche in grado di interpretare l’importante azione catalitica che alcuni acidi (come AlCl3) svolgono in alcune reazioni organiche (per es., in quelle di alchilazione). 1.1 Forza di un a. La definizione di Brönsted di acido e base coniugati può essere schematizzata mediante l’equilibrio:
[1] acido 1 + base 2 ⇄ acido 2 + base 1,
dove acido 1/base 1 e acido 2/base 2 sono le due coppie di acidi e basi coniugati. Nel caso della dissociazione di un acido in acqua, la [1] diventa:
[2] acido + H2O ⇄ H3O+ + base.
L’equilibrio [2] è caratterizzato dalla costante di equilibrio termodinamico K espressa da:
[3] formula
dove i termini al secondo membro rappresentano le attività delle diverse specie. Poiché l’attività di una specie è data dal prodotto della concentrazione C per il coefficiente di attività f si ottiene:
[4] formula

Per soluzioni molto diluite i coefficienti di attività tendono a uno mentre l’attività della specie acqua diviene praticamente costante. In tali condizioni si ha perciò:
[5] formula
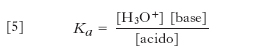
dove Ka è detta costante di acidità. Per es., nel caso dell’acido acetico si ha:
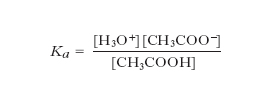
Il rapporto [acido]/[base] dipende dal valore di Ka, che pertanto può essere utilizzato per valutare la forza di un acido. I suoi valori per alcuni dei più comuni acidi sono riassunti nella tabella. La base coniugata di un acido debole è in grado di dare reazione di idrolisi con l’acqua con costante basica data da:
[6] formula
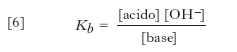

Combinando la [6] con la [5] e tenendo presente il prodotto ionico dell’acqua Kw = [H3O+] [OH−], si ha che Kb=Kw/Ka. Dal fatto che i valori di Ka e di Kb sono inversamente proporzionali, discende che un acido è tanto più forte quanto più debole è la sua base coniugata e viceversa. Dalla definizione di Brönsted risulta inoltre che la forza di un acido dipende dalla natura del solvente utilizzato. L’acqua è un solvente dalle spiccate proprietà protolitiche, sicché molti acidi risultano completamente dissociati in acqua e dunque appaiono caratterizzati dalla stessa forza (effetto livellante dell’acqua); invece, in un solvente meno basico dell’acqua, i medesimi acidi possono mostrare diversa forza. Un vantaggio della definizione di acido di Brönsted è che, per un dato solvente, è possibile costruire una scala univoca della forza degli acidi; inoltre, l’ordine di tale scala non muta sostanzialmente con il solvente. La teoria di Lewis è invece meno utile in questo contesto, perché le forze relative degli acidi variano notevolmente a seconda della base di riferimento.


