Chimica bioinorganica
Enciclopedia Italiana - VII Appendice (2006)
Chimica bioinorganica
di Ivano Bertini e Paola Turano
La c. b. (o chimica inorganica biologica) si occupa della chimica degli ioni metallici nei sistemi biologici. Carbonio, idrogeno, azoto, ossigeno, fosforo e zolfo sono i costituenti di tutti i componenti della cellula: proteine, acidi nucleici, lipidi, membrane, zuccheri, metaboliti. La chimica di questi sei elementi e dei loro composti è oggetto della biochimica classica. Tuttavia, perché ci sia vita è necessaria la presenza di un'altra ventina di elementi, storicamente considerati non-bio e quindi inorganici.
Il nome di questa disciplina riflette il fatto che la comprensione a livello atomico di sistemi coinvolgenti elementi inorganici all'interno di matrici biologiche (solitamente proteine o acidi nucleici) richiede competenze che vanno dalla chimica inorganica alla biologia.
Introduzione storica
J.J. Berzelius, negli anni 1837-38, individuò i pigmenti nelle foglie. Nel 1869 F. Hoppe-Seyler identificò il pigmento del sangue cui dette il nome di emoglobina. Nei primi anni del Novecento C.A. Schunck e L.P. Marchlewski ebbero il merito di riconoscere analogie chimiche fra le due classi di pigmenti. Tuttavia soltanto con R.M. Willstätter fu dimostrato come il magnesio fosse un componente fondamentale della clorofilla. Nel 1915, durante la consegna del premio Nobel per la chimica a Willstätter, il presidente del Comitato del Nobel per la chimica sottolineò il ruolo del novello laureato nel dimostrare che il magnesio era parte integrante della clorofilla così come il ferro lo era dell'emoglobina. Nel 1897 K.A.H. Mörner identificò per via spettroscopica il miocromo, in seguito chiamato mioglobina. Tra il 1880 e il 1890 C.A. MacMunn osservò, denominandoli istoematine o mioematine, i pigmenti che, dopo un periodo di oblio, furono riscoperti da D. Keilin a Cambridge nel 1925, e rinominati citocromi. Il lavoro Del citocromo, un pigmento respiratorio comune a animali, lievito e piante superiori, pubblicato da Keilin nel 1925 nei Proceedings of the Royal Society, segnò l'inizio degli studi su quella che in seguito O.H. Warburg definì catena respiratoria. Nel 1936 Keilin riuscì a purificare il citocromo c con una purezza di circa l'80%, e vicina al 100% nel 1939. Nel 1941 H. Theorell e Å. Åkeson identificarono nella catena laterale imidazolica di un'istidina uno dei leganti assiali del citocromo c. Sempre nei primi anni del Novecento H. Fischer condusse studi volti alla comprensione della natura chimica di clorofilla ed emina, che culminarono nella sintesi per via chimica di quest'ultima. Per tali risultati nel 1930 ricevette il premio Nobel per la chimica.
Gli studi di Keilin sono davvero fondamentali nel settore. A lui si deve anche la scoperta nella catena respiratoria mitocondriale di due tipi di citocromo, che lui chiamò a e a3, di un secondo citocromo c che chiamò c1, e infine di un ulteriore componente chiamato citocromo b. Come ricordò nella sua comunicazione alla cerimonia dei premi Nobel P.D. Mitchell, insignito del riconoscimento per la chimica nel 1978 per il suo contributo alla comprensione del trasferimento di energia in biologia, la visione semplice di Keilin della catena respiratoria si rivelerà completamente corretta. Nonostante questo, O.H. Warburg, premio Nobel per la medicina nel 1931 per la scoperta della natura e del modo di azione dell'enzima respiratorio, come si legge nella motivazione ufficiale, si rifiutò di riconoscere il ruolo dei citocromi. La diatriba scientifica fra i due costituì materia di controversia per tutti gli anni Trenta del 20° secolo.
In quel periodo molti iniziarono a interessarsi alla respirazione aerobica. Il termine ossidasi era stato introdotto da G. Bertrand nel 19° sec. per descrivere l'enzima contenente rame, oggi noto come laccasi. Nel periodo 1910-1920 F. Battelli e L. Stern studiarono estensivamente l'ossidazione di un certo numero di sostanze da parte dell'ossigeno in presenza di tessuti animali e dimostrarono la sensibilità del processo al cianuro, attribuendola a un enzima che chiamarono indofenolo ossidasi. A Keilin e a E.F. Hartree si deve anche la scoperta, nel 1939, della presenza di rame nell'enzima oggi conosciuto come citocromo c ossidasi, e che loro identificarono con l'indofenolo ossidasi. In quegli stessi anni sorsero due diverse scuole di pensiero sul meccanismo dell'ossidazione biologica: O. Wieland e T. Thunberg proponevano che il primo passo dell'ossidazione fosse costituito dalldell'idrogeno attraverso un processo catalizzato da deidrogenasi; Warburg, invece, sosteneva che il processo fondamentale fosse costituito dall'attivazione dell'ossigeno da parte di un enzima respiratorio contenente ferro (l'atmungsferment). Spettò ad A. Szent-György (premio Nobel per la medicina nel 1937) conciliare le due visioni dimostrando che i due processi sono tra loro complementari e che nelle cellule del muscolo l'ossigeno attivato attiva l'idrogeno. In riconoscimento del ruolo dei citocromi di Keilin, l'enzima respiratorio fu definitivamente chiamato citocromo ossidasi.
La storia della citocromo c ossidasi è emblematica per dimostrare come negli anni che vanno dalla fine del 19° sec. all'inizio del 20°, tanta parte degli studi chimici e medico-fisiologici fosse incentrata sull'identificazione di funzioni enzimatiche. Risale ancora a Berzelius il concetto di catalizzatore biologico che lui definì come forza catalitica del corpo. Nel 1926 Willstätter riconobbe la sua incapacità a identificare la natura chimica degli enzimi, dopo innumerevoli tentativi di produrli in forma pura. Nello stesso anno il ricercatore americano J.B. Sumner pubblicò un lavoro in cui riportò la cristallizzazione in forma pura dell'enzima ureasi (che oggi sappiamo contenere nichel), capace di decomporre l'urea in biossido di carbonio e ammoniaca. La scoperta di Sumner che l'enzima altro non era che una proteina, fu tuttavia dibattuta a lungo e solo lentamente accettata (infatti, ci vollero vent'anni prima che gli venisse assegnato il premio Nobel per la chimica per tale scoperta). Gli studi di enzimologia portarono anche alla scoperta di altri enzimi, in seguito risultati essere zinco enzimi: è questo il caso dell'anidrasi carbonica identificata da N.U. Meldrum e F.J.W. Roughton nel 1933; dell'alcol deidrogenasi, purificata da lievito e cristallizzata per la prima volta nel 1936 da E. Negelein e H.J. Wulf; della carbossipeptidasi identificata da M.L. Anson nel 1937. Nonostante il ruolo dello zinco come essenziale per la sopravvivenza di alcuni organismi fosse noto già dalla seconda metà del 19° sec., bisogna arrivare a B.L. Vallee negli anni Cinquanta del 20° sec. perché la presenza di zinco sia rivelata come essenziale per la funzione catalitica. Ancora una volta è necessario menzionare Keilin che, insieme a T. Mann, nel 1940 riportò sull'ubiquità dello zinco negli organi umani.
Nel 1941 H. Theorell e i suoi collaboratori cristallizzarono la perossidasi da rafano e dimostrarono che era costituita, oltre che da una catena polipeptidica, da carboidrati ed emina (e quindi ferro). Nel 1955 Theorell fu insignito del premio Nobel per la medicina, per le sue scoperte sulla natura e il modo di azione degli enzimi ossidativi.
Nel 1960 M.F. Perutz pubblicò la struttura a raggi X dell'emoglobina. Nello stesso anno J.C. Kendrew pubblicò la struttura a raggi X della mioglobina. Per questi risultati furono insigniti del premio Nobel per la chimica nel 1962. È l'inizio di una nuova era. D'ora in poi la conoscenza della struttura tridimensionale di ogni macromolecola diventerà centrale per la comprensione della sua funzione. Due anni dopo, il Nobel per la chimica venne assegnato a D. Crowfoot Hodgkin, ancora per studi strutturali su importanti sostanze biochimiche, tra cui la vitamina B12 che contiene cobalto.
La c. b. ha cominciato ad affermarsi a livello mondiale con un'identità propria tra la fine degli anni Settanta e l'inizio degli anni Ottanta del 20° sec., ed è ancora in espansione.
Ioni metallici e loro ruolo
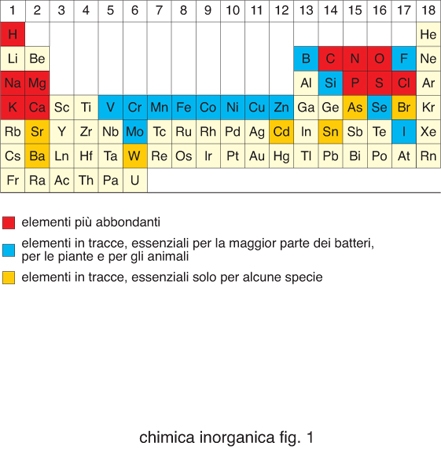
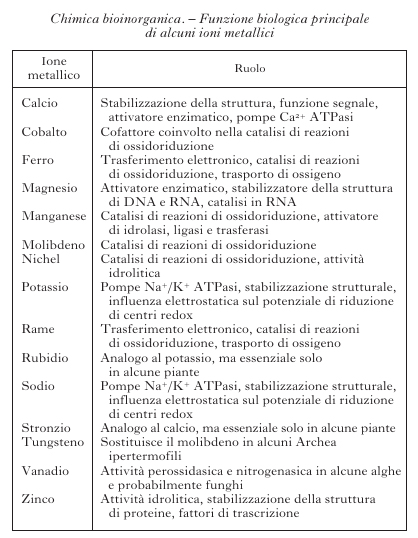
Degli 80 elementi non radioattivi della tavola periodica (fig. 1), poco meno di metà sono importanti per i sistemi viventi. Cloro, sodio, potassio, magnesio e calcio, così come ferro, rame e zinco, sono necessari per tutte le forme di vita e quindi essenziali. Boro, fluoro, vanadio, cromo, manganese, cobalto, nichel, molibdeno, silicio, selenio e iodio sono necessari per la maggior parte delle specie viventi, mentre arsenico, bromo, stronzio, bario, cadmio, stagno e tungsteno sono utilizzati soltanto da un numero limitato di organismi. Gli elementi inorganici, in genere, e gli ioni di elementi metallici, in particolare, svolgono funzioni molto disparate all'interno dei sistemi biologici. La tabella riporta un sommario dei principali ruoli da essi svolti. La presenza di determinati ioni metallici nei sistemi biologici ha una determinante chimica e una ambientale. Gli ioni metallici possiedono proprietà chimiche quali potenziali di riduzione, chimica di coordinazione, proprietà acido/base di Lewis ecc., che sono uniche all'interno della tavola periodica e diverse tra loro. Questi ioni diventano quindi essenziali per lo svolgimento di determinate reazioni metaboliche. Tuttavia, la selezione di certi ioni metallici rispetto ad altri nasce anche dal fatto che la vita si è sviluppata in presenza dei minerali della crosta terrestre e degli ioni disciolti nelle acque del nostro pianeta. La biodisponibilità di ciascun elemento è stata quindi altrettanto importante per definirne la distribuzione nei sistemi viventi. La biodisponibilità cambia da un ambiente a un altro e questo fa sì che organismi estremofili possano utilizzare come essenziali elementi che non lo sono per la maggior parte delle specie viventi.
Allo stesso tempo la biodisponibilità di determinati elementi è stata soggetta all'evoluzione del pianeta e in particolare, il passaggio da condizioni anaerobiche a quelle aerobiche ha determinato una grande rivoluzione, favorendo l'evoluzione delle specie preesistenti da anaerobiche in aerobiche o la migrazione di forme di vita anaerobica verso zone difficilmente accessibili all'ossigeno atmosferico.
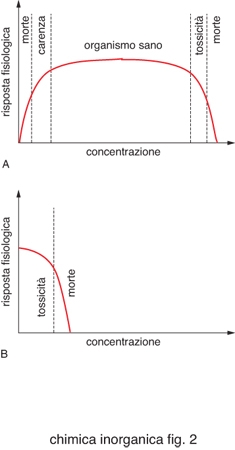
Per ogni elemento in un determinato organismo esistono dei valori ottimali di concentrazione, come schematizzato in fig. 2. Per i metalli essenziali esiste un intervallo abbastanza ampio di concentrazione che corrisponde allo stato di salute dell'organismo. Al di sotto di questo la quantità di metallo è insufficiente per le funzioni vitali a cui dovrebbe partecipare, al di sopra diventa tossico. Esistono invece metalli che non rientrano fra quelli evidenziati nella tavola periodica di fig. 1 e che sono tossici.
Gli esseri viventi devono difendersi ed eventualmente detossificarsi da questi ioni metallici o da metalli essenziali in eccesso. Tuttavia, un metallo tossico può essere tollerato a basse dosi. Su questo principio si basa l'uso di farmaci contenenti ioni metallici, in quell'aspetto peculiare della c. b. definito metalli in medicina. L'utilizzo di composti inorganici come farmaci è ben documentato seppur spesso empirico.
Nella sua associazione funzionale con una proteina o un acido nucleico, lo ione metallico viene chiamato cofattore metallico.
I cationi metallici possono interagire con la parte esposta della doppia elica del DNA, che può essere vista come un filamento lineare di carica negativa dovuta alla presenza di gruppi fosfato esposti al solvente. Per l'RNA la situazione è meno semplice, perché la presenza di struttura terziaria crea un potenziale elettrostatico irregolare, con gruppi fosfato che vengono a trovarsi raggruppati in certe zone della struttura. Di conseguenza i cationi metallici possiedono un'affinità particolarmente elevata per queste zone ad alta densità di carica negativa. L'effetto è sinergico: gli ioni metallici hanno un ruolo importante per la stabilità del fold della molecola di RNA e la loro presenza facilita il processo di folding. L'effetto stabilizzante è tanto maggiore quanto più alta è la carica dello ione e tanto più piccole sono le sue dimensioni. Il Mg2+ risulta essere lo ione preferito. L'RNA è capace di svolgere funzioni enzimatiche, catalizzando un certo numero di reazioni che vanno dalla semplice formazione del legame peptidico, che caratterizza la funzione dell'rRNA, a reazioni di self-cleavage e transesterificazioni del fosfodiestere. La presenza di alcuni ioni metallici si sta dimostrando fondamentale per la funzione catalitica.
Nonostante ci sia interesse per le interazioni ione metallico-acidi nucleici, la parte principale della c. b. è incentrata sullo studio delle interazioni ione metallico-proteina.
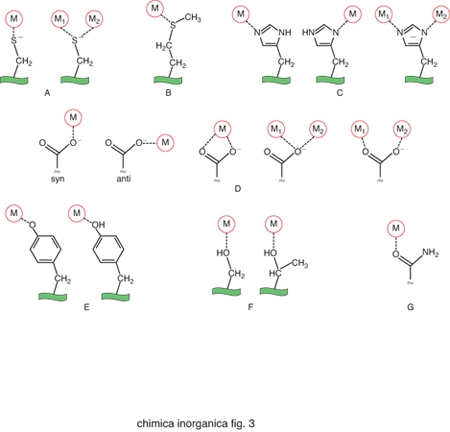
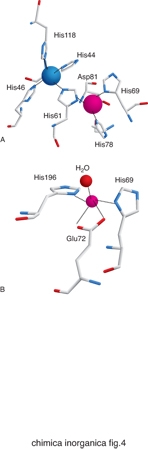
Gli amminoacidi che costituiscono le proteine non sono sufficienti da soli a dare luogo alle reazioni indispensabili per la vita dei vari organismi. Gli ioni metallici, quando associati alle catene polipeptidiche delle proteine, possono invece dare luogo a un'enorme varietà di reazioni o altre funzioni fisiologicamente importanti. La capacità delle proteine di legare ioni metallici deriva dalle proprietà chimiche di un certo numero di catene laterali. Gli atomi di zolfo di cisteina e metionina, gli atomi di azoto dell'anello imidazolico dell'istidina e gli atomi di ossigeno del glutammato, dell'aspartato e della tirosina si comportano come atomi donatori nei confronti degli ioni metallici, dando luogo a legami di coordinazione. Pertanto questi aminoacidi rappresentano i più comuni leganti dei cofattori metallici. In alcuni casi gli atomi donatori possono anche essere l'ossigeno del carbonile peptidico oppure delle catene laterali di serina e treonina, o l'atomo di azoto del gruppo ammidico del legame peptidico o della catena laterale della lisina. La fig. 3 riassume schematicamente le modalità di coordinazione possibili per ciascuno di questi atomi donatori. Nella fig. 4 sono invece riportati i siti metallici di due proteine scelte come esempio per illustrare l'aspetto dell'intorno di coordinazione degli ioni metallici.
Quando l'affinità fra lo ione metallico e la proteina è al di sopra di un certo valore di soglia si parla di metalloproteina. Gli enzimi metallo-dipendenti vengono detti metalloenzimi.

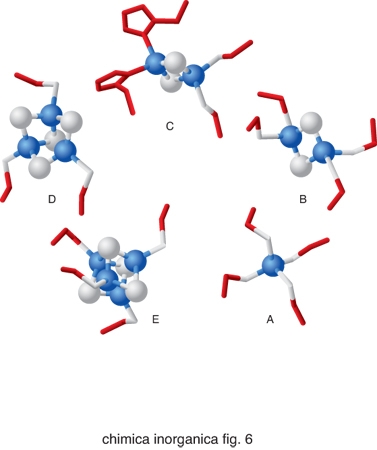
Nonostante la grande versatilità in termini di reattività dei cofattori metallici sopra descritti, essi non sono sufficienti a coprire tutte le possibili necessità biologiche. La natura ricorre anche a una strategia alternativa alla semplice incorporazione dello ione metallico all'interno di proteine, utilizzando strutture più complesse. Si parla in questo caso di cofattori metallici speciali. Esempi fra i più diffusi di costrutti utilizzati in cofattori metallici speciali sono gli anelli tetrapirrolici (fig. 5). Essi sono costituiti dalle porfirine che legano lo ione ferro, dando luogo alle cosiddette eme-proteine, dalla clorofilla, nella quale l'anello tetrapirrolico è invariabilmente legato al catione Mg2+, e dalla vitamina B12, in cui l'anello corrinico coordina lo ione Co2+. Nelle eme-proteine lo ione ferro coordinato alla porfirina completa la sua coordinazione legandosi a 1 oppure 2 atomi donatori di catene laterali della proteina e/o molecole di H2O. La sua reattività dipende enormemente dalla natura di questi leganti assiali. Un altro cofattore metallico speciale molto comune è il cluster ferro-zolfo, in cui gli atomi di ferro sono coordinati sia ad atomi di zolfo di catene laterali di cisteina sia ad anioni S2−. I cluster ferro-zolfo più comuni sono illustrati in fig. 6. Quello di fig. 6A non è propriamente un cluster, contenendo solo uno ione ferro legato a 4 cisteine, ma può essere considerato il precursore delle altre strutture più complesse. Il cluster di fig. 6B contiene 2 ioni ferro, 2 anioni S2− e 4 cisteine; il cluster di fig. 6C è analogo ma ci sono 2 istidine al posto di 2 cisteine; il cluster della fig. 6E è il cosiddetto cubano, con 4 ioni ferro, 4 anioni S2− e 4 cisteine; infine il cluster della fig. 6D è analogo ma manca uno dei 4 ioni ferro.
Zinco, ferro e rame
A partire dagli ultimi anni del 20° sec., la disponibilità del genoma di un numero crescente di organismi ha operato una rivoluzione nel campo delle scienze biologiche molecolari, consentendo la possibilità di identificare tutte le proteine che i singoli organismi possono produrre. Tuttavia, l'identificazione delle metalloproteine non è ovvia. La sequenza dei geni è associata alla sequenza amminoacidica della proteina. La difficoltà sta nel prevedere se una certa proteina lega uno o più ioni metallici per svolgere la sua funzione. Tuttavia esistono protocolli bioinformatici allo scopo di individuare nuove potenziali metalloproteine sulla base del confronto di nuove sequenze con pattern tipici per i leganti dello ione metallico in metalloproteine già note.
Lo zinco
Questo tipo di approccio è stato, per es., utilizzato per identificare le zinco proteine codificate dal genoma umano. Lo zinco è un elemento essenziale e in termini di abbondanza fra gli ioni metallici di transizione negli organismi viventi è secondo soltanto al ferro. Come indicato in tabella 1, le funzioni principali dello zinco nei sistemi viventi sono tre. Lo zinco è spesso trovato in siti in cui è tetracoordinato a catene laterali di aminoacidi per conferire stabilità strutturale alla proteina. Un esempio di zinco strutturale è rappresentato dalla proteina Cu,Zn superossido dismutasi, dove la presenza di Zn2+ legato a tre istidine e un aspartato è fondamentale per la stabilità strutturale dell'enzima, la cui attività dipende invece dalla presenza dello ione rame. Un esempio particolare di zinco strutturale è rappresentato dal caso delle proteine chiamate zinc-finger. Si tratta di piccoli domini proteici tipici dei fattori di trascrizione o di altre proteine che legano il DNA. Essenziale per la forma a dito tipica di ciascun dominio è la presenza di uno ione Zn2+, che risulta coordinato a quattro catene laterali di istidine o cisteine in intorni di tipo tetraedrico. Pattern di tre leganti aminoacidici del tipo His, His, His o His, Asp, His sono tipici dello ione zinco in zinco enzimi. Un esempio di coordinazione His, Asp, His è esemplificato dalla fig. 4B per l'enzima carbossipeptidasi. In generale, l'attività principale degli zinco enzimi riguarda l'attivazione di una molecola di acqua (che va a coordinare lo ione zinco in aggiunta ai tre leganti aminoacidici). L'acqua così attivata diventa capace di fare un attacco nucleofilo sul substrato. I substrati possibili sono i più vari (legami peptidici, collagene, fosfolipidi). Per quanto queste proprietà strutturali e di reattività fossero note, la novità insita nell'approccio bioinformatico basato sull'analisi delle banche dati genomiche consiste nella capacità di contare e catalogare, sulla base dei pattern di leganti il metallo, le potenziali zinco proteine ancora ignote. A testimonianza del ruolo biologico fondamentale di questo ione basta considerare che l'analisi del genoma umano ha consentito di identificare 2800 zinco proteine putative, che corrispondono al 10% dell'intero proteoma umano. Di queste, il 40% può essere assegnato a fattori di trascrizione e circa il 50% a zinco enzimi. Un'analisi analoga dei genomi di tutti gli organismi disponibili a oggi indica diverse percentuali di distribuzione per le varie zinco proteine putative.
Nei batteri le zinco proteine putative sono in media un centinaio, ossia rappresentano soltanto il 3-4% del proteoma totale, e il 63% è costituito da zinco enzimi. Negli Archea le zinco proteine putative sono in media un'ottantina, ossia circa il 4% del proteoma totale, e il 48% è costituito da zinco enzimi. Negli altri eucarioti la situazione non è troppo diversa dal caso del genoma umano.
Il ferro
Il ferro è uno degli elementi più abbondanti della crosta terrestre e di conseguenza viene utilizzato sostanzialmente da tutte le specie viventi per le funzioni più disparate. Il ferro rappresenta infatti l'elemento di transizione più abbondante nei sistemi viventi. Esso è presente come Fe2+ e Fe3+. La forma a stato di ossidazione +3, favorita in condizioni aerobiche, risulta insolubile per la formazione di idrossidi, essendo quindi poco biodisponibile. Per questi motivi gli organismi viventi hanno sviluppato dei sistemi di solubilizzazione, sequestro, trasporto e rilascio del ferro. Nei vertebrati, le proteine coinvolte sono lattoferrina, transferrina e ferritina. I batteri si procurano il ferro attraverso sistemi di acquisizione differenziati. Possono utilizzare piccole molecole chelanti, dette siderofori, o avere recettori di membrana esposti e capaci di legare proteine di trasporto del ferro di altri organismi. Il ferro nei sistemi viventi si trova inoltre sequestrato in cofattori speciali quali i gruppi eme e i cluster ferro-zolfo. La biosintesi dell'eme e il suo inserimento nella matrice proteica sono generalmente processi controllati da una serie di altre proteine codificate dallo stesso organismo. L'eme libero è di per sé tossico in quanto l'anello tetrapirrolico costituisce un'entità idrofobica che può andare a inserirsi nelle membrane cellulari e la presenza dello ione ferro può promuovere reazioni di ossidoriduzione indesiderate. La protezione dall'eme libero è assicurata dall'emopexina. Nei batteri gram-negativi esistono proteine extracellulari, dette emofori, che possiedono un'alta affinità per l'eme legato alle eme proteine dell'organismo ospite (mioglobina, emoglobina, emopexina). La loro funzione è quella di sequestrare l'eme e di rilasciarlo a specifici recettori di membrana del batterio gram-negativo.
Da un punto di vista funzionale le eme proteine svolgono le funzioni più disparate, che possono tuttavia essere riassunte in quattro classi principali: trasporto dell'ossigeno, reazioni di trasferimento elettronico, catalisi di reazioni di ossidoriduzione, funzione segnale. I trasportatori di ossigeno sono la mioglobina e l'emoglobina, che sono state descritte nella parte storica in quanto tra le prime metalloproteine identificate e le prime due per le quali sia stata risolta la struttura tridimensinonale. In queste proteine il ferro è legato ai quattro atomi di azoto dell'anello porfirinico e all'azoto imidazolico di una istidina. La sesta posizione di coordinazione rimane accessibile per il legame della molecola di O2. I citocromi sono invece eme proteine in cui il ferro è coordinato all'anello della porfirina e a due leganti assiali forniti da catene laterali di aminoacidi (solitamente due istidine o una istidina e una metionina). Lo ione ferro cicla tra i due stati di ossidazione Fe2+/Fe3+ trasportando così un elettrone da una proteina donatore a una proteina accettore. L'accettore è spesso un metalloenzima che ha la funzione di catalizzare reazioni di riduzione, in cui l'elettrone acquisito dal citocromo viene trasferito a una molecola, substrato dell'enzima. Un esempio tipico è il citocromo c mitocondriale che svolge il ruolo di trasportatore di elettroni all'interno della catena respiratoria. In questo caso l'accettore di elettroni è costituito dalla proteina citocromo c ossidasi, che riduce l'O2 a H2O. Gli eme enzimi hanno tipicamente la funzione di catalizzatori di reazioni di ossidoriduzione. Tali reazioni possono essere le più disparate in termini di substrato: dalla citata riduzione dell'ossigeno molecolare ad acqua, all'ossidazione di ammoniaca a nitrito, alla riduzione di nitrito ad ammonio, e così via. Di fatto eme enzimi si ritrovano fra le proteine preposte alle reazioni di ossidoriduzione tipiche dei cicli biogeochimici di ossigeno, azoto, carbonio. In tutti questi enzimi lo ione ferro è tipicamente pentacoordinato o esacoordinato con un legante assiale debole nella sesta posizione di coordinazione. In tempi più recenti è stato dimostrato inoltre che alcune eme proteine con ferro pentacoordinato sono capaci di legare piccole molecole, quali NO, CO, comportandosi come sensori per lo stress ossidativo o messaggeri ed esercitando quindi una funzione segnale.
Le ferro proteine contenenti cluster ferro-zolfo svolgono tipicamente la funzione di trasportatori di elettroni, analogamente ai citocromi. Tuttavia esistono anche cluster ferro-zolfo con funzione di sensori di O2, NO, O2− o con attività catalitica.
Oltre alle proteine di trasporto del ferro, esistono infine numerose altre ferro proteine non-eme non-ferro-zolfo. In queste proteine il ferro è solitamente legato ad atomi di azoto o di ossigeno, in particolare di catene laterali di istidine, glutammati e aspartati. Il ferro può essere presente in centri mononucleari o binucleari. Questo tipo di siti ha normalmente una funzione catalitica, e la reattività principale è associata all'attivazione dell'O2 e alla sua inserzione in legami C-H mediata dai ferro-enzimi mono- e diossigenasi.
L'analisi bioinformatica del genoma di 57 organismi (12 Archea, 40 batteri, 5 eucarioti, incluso l'Homo sapiens) per l'identificazione di potenziali ferro proteine non-eme indica che la percentuale di FeS diminuisce sensibilmente nell'ordine Archea, batteri, eucarioti.
Il rame
La maggior parte dei cofattori biologici a rame svolge una delle seguenti funzioni: trasferimento elettronico, catalisi di reazioni di ossidoriduzione, trasporto di ossigeno. La funzione di trasporto elettronico avviene essenzialmente attraverso le cosiddette proteine blu di rame, in cui lo ione rame cicla fra gli stati di ossidazione +2 e +1 ed è coordinato in un intorno piramidale trigonale a una cisteina, a due istidine e a un quarto legante più debole (tipicamente lo zolfo di una metionina). Si parla in questo caso di rame di tipo 1. Tuttavia esistono centri di nucleari di rame (il cosiddetto CuA) in cui due ioni rame sono ciascuno tetracoordinato, con due cisteine a ponte tra i due ioni metallici. Questi centri sono preposti al trasferimento elettronico intramolecolare e sono presenti nelle ossidasi (per es., la citocromo c ossidasi); il cofattore cicla formalmente tra gli stati di ossidazione [Cu2+Cu+] e [Cu2+]. Esistono altri due tipi di cofattori a rame. Il cosiddetto rame di tipo 2, che è un centro mononucleare di rame, in cui lo ione metallico ha 4-5 leganti, nessuno dei quali contiene atomi di zolfo come donatori. Centri di tipo due sono tipici degli enzimi con attività di catalizzatori di reazioni di ossidoriduzione. Il rame di tipo 3 è invece costituito da due ioni rame, ciascuno legato a tre catene laterali di istidine. A ponte tra i due ioni rame possono legarsi piccole molecole o ioni, quali O2, OH−. Questo schema è comune sia a enzimi che catalizzano reazioni di ossidoriduzione, quali le tirosinasi, sia a proteine di trasporto dell'ossigeno, come l'emocianina (tipica dei molluschi e degli artropodi).
Sono emerse tuttavia nuove proteine capaci di legare il rame, la cui funzione è essenzialmente quella di importarlo nella cellula, sequestrarlo e indirizzarlo nei vari comparti cellulari e alle proteine ed enzimi che contengono cofattori rame essenziali per la loro funzione. Dalla metà degli anni Novanta del 20° sec., è infatti dimostrato che il livello di rame libero all'interno della cellula è estremamente basso, essendo lo ione sostanzialmente sequestrato all'interno di proteine preposte specificatamente al suo trasporto. All'interno della cellula sono quindi presenti veri e propri percorsi di trasporto controllato del rame, che differiscono in numero di proteine coinvolte e complessità a secondo dell'organismo. Nel caso del lievito Saccharomyces cerevisiae sono state individuate le proteine di trasporto verso il mitocondrio per l'assemblaggio dei centri rame della citocromo c ossidasi e per l'assemblaggio nel citosol della Cu,Zn superossidodismutasi. Simili meccanismi di trasporto sono stati individuati anche nell'uomo e in alcuni batteri. Lo studio dell'omeostasi dei metalli rappresenta una frontiera della ricerca bioinorganica.
Bibliografia
J.J.R. Frausto da Silva, R.J.P. Williams, The natural selection of the chemical elements: the environment and life's chemistry, Oxford 1996.
Handbook on metalloproteins, ed. I. Bertini, A. Sigel, H. Sigel, New York-Basel 2001.
Biological inorganic chemistry: structure & reactivity, ed. I. Bertini, H.B. Gray, E.I. Stiefel, J. Selverstone Valentine, Mill Valley (CA) 2006.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


