Elettrochimica
Enciclopedia della Scienza e della Tecnica (2008)
Elettrochimica
L'elettrochimica ebbe origine agli inizi del XIX secolo. È convenzione indicarne l'atto di nascita con la lettera del 20 marzo 1800 di Alessandro Volta a Joseph Banks, presidente della Royal Society di Londra, intitolata On the electricity excited by mere contact of conducting substances of different kinds, nella quale Volta descriveva la generazione di elettricità da parte di un apparato elettromotore, che sarebbe stato poi denominato pila di Volta. Questa consisteva in una pila di coppie di dischi di zinco e di rame (o di zinco e di argento), intramezzate da dischi di cartone imbevuto di soluzione salmastra. Solo pochi mesi dopo William Nicholson e Anthony Carlisle usarono tale apparato per generare elettricità, con la quale decomposero l'acqua nei suoi elementi, ossia l'idrogeno e l'ossigeno. È di questo processo a due vie ‒ la produzione di elettricità da una reazione chimica e quella di modificazioni chimiche mediante il passaggio di corrente elettrica ‒ che si occupa sostanzialmente l'elettrochimica, sebbene le sue ramificazioni nella scienza moderna siano molto ampie.
L'invenzione di Volta fu il culmine di alcuni decenni di ricerche sull'elettricità animale, cioè sulla capacità di certe specie marine, come la torpedine e l'anguilla elettrica, di produrre scosse elettriche. Questo fenomeno suscitava grande interesse, ma fu l'osservazione di Luigi Galvani (i cui risultati furono pubblicati nel 1791), dell'eccitazione elettrica di zampe di rana isolate dal corpo, che spinse Volta alla ricerca che avrebbe portato all'invenzione della pila. In breve, Galvani sosteneva che l'elettricità animale era un fenomeno puramente biologico, mentre Volta affermava che essa era essenzialmente identica all'elettricità statica (da strofinio) e dimostrò che poteva essere generata senza la partecipazione di sistemi viventi. Le ricerche successive hanno confermato la conclusione di Volta, ma hanno anche dato ragione dell'importante ruolo dei processi elettrochimici nei sistemi biologici.
È possibile che processi di tipo elettrochimico fossero usati empiricamente anche nell'Antichità. Scavi effettuati nel 1930 vicino a Baghdad hanno portato alla luce vasi di argilla contenenti bacchette di ferro, risalenti al II sec. a.C., che si pensa siano una sorta di batterie primitive. È stato ipotizzato che tali batterie potessero essere utilizzate per la doratura elettrolitica. Il termine elettrochimica fu introdotto da Humphry Davy nel 1807, a conclusione di uno straordinario periodo di attività presso la Royal Institution a Londra, durante il quale egli fornì solide basi alla nuova disciplina. In particolare, Davy utilizzò metodi elettrochimici per scoprire nuovi elementi: potassio, sodio, calcio, stronzio, bario, berillio e alluminio. Il suo assistente, Michael Faraday, dimostrò poi l'identità di tutte le forme di elettricità e trovò la relazione quantitativa fra la quantità di elettricità passata attraverso una cella elettrolitica e la quantità di sostanza modificata chimicamente (leggi di Faraday). Oltre a queste eleganti ricerche sperimentali, Faraday propose la nomenclatura elettrochimica usata ancora oggi: elettrolisi, elettrodo, elettrolita, catodo, anodo, anione, catione, e così via.
Celle elettrochimiche
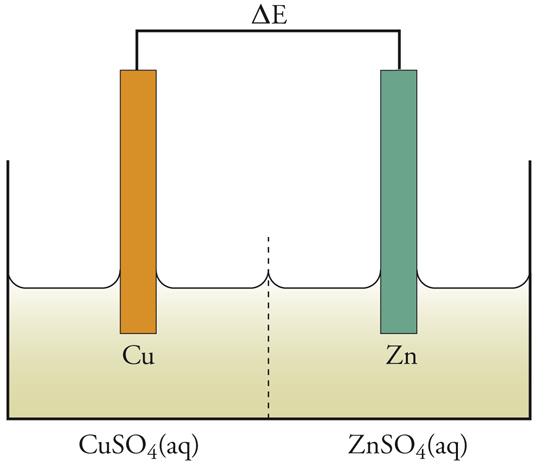
La pila di Volta era un insieme di unità, ognuna delle quali consisteva in due dischi di metalli diversi, separati da un disco di cartone imbevuto di soluzione salmastra. Questo è un esempio di cella elettrochimica, che ha sempre due elettrodi. Un prototipo semplice di cella, infatti, potrebbe essere l'elettrodo di rame combinato con uno di zinco (fig. 2). Le reazioni che avvengono a questi elettrodi sono le seguenti:
[1] Cu2+(aq)+2e− ←→ Cu
[2] Zn2+(aq)+2e− ←→ Zn.
È la diversa reattività dei due metalli, Cu e Zn, con l'elettrolita a fornire la differenza di potenziale che fa passare la corrente elettrica attraverso la cella. La combinazione delle reazioni [1] e [2] nella forma:
[3] Zn+Cu2+(aq) → Cu+Zn2+(aq)
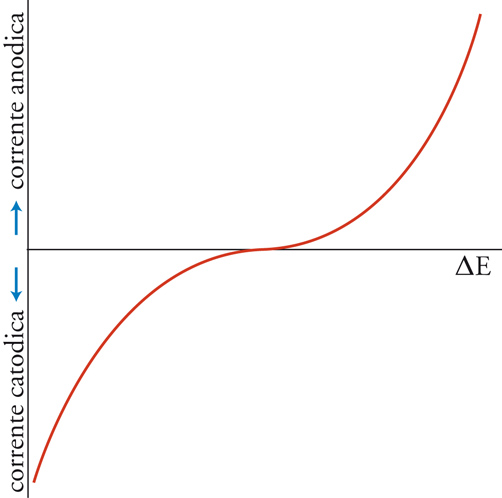
dà la reazione globale nella cella, che produce elettricità poiché è una reazione spontanea. Se però alla cella viene applicata una differenza di potenziale esterna (ΔE) sufficientemente grande, la reazione può essere invertita; cioè la reazione chimica procederà in direzione opposta a quella spontanea mostrata nella [3], come risultato del passaggio forzato della corrente elettrica. Ciò può essere rappresentato mediante un diagramma del flusso di corrente in funzione del potenziale applicato esternamente (fig. 3). Per un particolare valore di ΔE, in corrispondenza del quale la corrente è 0, la reazione nella cella è bilanciata dalla differenza di potenziale esterna. Questa ΔE è una grandezza caratteristica della cella, nota come f.e.m. (forza elettromotrice), termine che ha perso il suo significato originario, sebbene la grandezza in sé sia di notevole importanza.
Hermann von Helmholtz e Josiah Willard Gibbs hanno dimostrato che la f.e.m. E di una cella in equilibrio è correlata alla variazione di energia libera di Gibbs ΔG della reazione che avviene nella cella mediante la relazione:
[4] formula
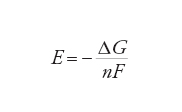
dove n è il numero di elettroni che attraversano il circuito esterno per ogni ione che reagisce all'elettrodo (nell'esempio di cui sopra n=2, come risulta dalle reazioni parziali [1] e [2]) e F è la costante di Faraday (96.484 C mol−1), che equivale alla carica di una mole di elettroni. La [4] è un'equazione fondamentale, poiché costituisce il ponte fra la termodinamica e le proprietà delle celle elettrochimiche in equilibrio. Queste proprietà possono essere interpretate in termini di reattività chimica. Uno dei numerosi risultati dell'applicazione della [4] è la deduzione di come la f.e.m. di una cella dipenda dalla concentrazione dei reagenti nella cella. L'energia libera di Gibbs, e di conseguenza la f.e.m., dipendono logaritmicamente dalle attività delle specie chimiche che partecipano a una reazione, secondo l'equazione:
[5] formula

dove R è la costante universale dei gas (8,31441 JK−1 mol−1), T è la temperatura termodinamica, ai è l'attività della specie i, ovvero la concentrazione attiva con la quale l'elettrolito prende parte agli equilibri che si stabiliscono in soluzione, e νi è il suo coefficiente stechiometrico nella reazione che avviene nella cella (i prodotti di reazione hanno valori positivi di νi e i reagenti valori negativi); E° è la f.e.m. quando tutti i reagenti hanno attività unitaria (ogni specie nel suo stato standard). La [5] è nota come equazione di Nernst.
Poiché le energie di Gibbs sono proprietà additive delle reazioni, anche la f.e.m. standard è una proprietà additiva. Perciò è possibile compilare una tabella delle f.e.m. standard delle celle in cui ogni elettrodo è collegato con un elettrodo standard e, di conseguenza, calcolare la f.e.m. di tutte le altre combinazioni. Per celle con elettroliti acquosi (e altri elettroliti in cui il protone solvatato è stabile), l'elettrodo standard è l'elettrodo a idrogeno, che consiste in un metallo cataliticamente attivo (generalmente platino), sul quale si fa gorgogliare idrogeno, immerso in una soluzione acida nella quale l'attività degli ioni idrogeno è unitaria.
Cinetica delle reazioni elettrodiche
Il fatto che la corrente passante attraverso la cella non diventi infinita quando il potenziale applicato esternamente differisce dalla f.e.m. di equilibrio, indica l'esistenza di processi lenti implicati nel passaggio di questa corrente. Nel metallo e nell'elettrolita questi processi sono espressi dalla resistenza elettrica dei materiali, causata, nel caso degli ioni, dal fatto che il loro movimento può avvenire solo spingendo 'da parte' altre particelle e, nel caso degli elettroni, dal moto termico del reticolo metallico, nonché dai difetti presenti in questo. Comunque, queste caratteristiche di resistenza non spiegano il comportamento di una cella elettrochimica al passaggio della corrente: essa mostra una resistenza maggiore, che è non lineare, nel senso che la corrente non è una funzione lineare del potenziale applicato, come sarebbe se gli unici effetti presenti fossero quelli della resistenza di massa. Gli effetti addizionali sono dovuti alla lentezza della stessa reazione elettrodica e al fatto che si tratta di una reazione eterogenea, nel senso che lo scambio dei portatori di carica avviene solo all'interfaccia. Formalmente, il tipo più semplice di reazione elettrodica è quello in cui il reagente e il prodotto sono costituiti da uno stesso ione in due diversi stati di ossidazione di uno stesso ione, entrambi presenti nell'elettrolita, e la reazione consiste nello scambio di un elettrone con l'elettrodo metallico. Un esempio di questo tipo di reazione potrebbe essere quello tra ione ferroso e ione ferrico:
[6] Fe2+(aq) ←→ Fe3+(aq)+e−.
La cinetica di questa reazione segue le leggi della normale cinetica chimica e, in particolare, è del primo ordine in entrambe le direzioni, verso destra (anodica) e verso sinistra (catodica), cioè la velocità di reazione r è proporzionale alla prima potenza della concentrazione del reagente:
[7] r→=k→[Fe2+]
[8] r←=k←[Fe3+]
dove [Fe2+] e [Fe3+] sono le concentrazioni locali rispettivamente di Fe2+(aq) e Fe3+(aq) nel luogo della reazione. La caratteristica delle equazioni di velocità delle reazioni elettrodiche è che le costanti di velocità k→ e k← dipendono dal potenziale dell'elettrodo: nel caso più semplice l'energia di attivazione è una funzione lineare del potenziale dell'elettrodo. Ciò conduce a una dipendenza esponenziale delle costanti di velocità dal potenziale E:
[9] formula[10]
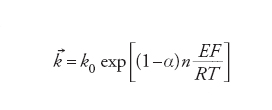
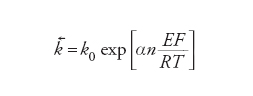
La costante α è detta coefficiente di trasferimento e il suo valore è normalmente compreso fra 0 e 1. Questa caratteristica unica delle reazioni elettrodiche significa che c'è un modo semplice per controllare la loro velocità e che, a causa della forma esponenziale, questo controllo può essere esercitato entro un intervallo molto ampio di velocità. La velocità netta della reazione, r→−r←, può essere espressa in termini di una corrente, poiché per ogni evento reattivo elementare n elettroni sono trasferiti al (o dal) metallo (n=1 nella reazione [6]). Questa è un'espressione delle leggi di Faraday. Poiché la reazione avviene all'interfaccia tra due fasi, la grandezza che interessa è la densità di corrente j e le velocità r→e r← sono espresse anche come velocità per unità di superficie dell'interfaccia. Si ha:
[11] j=nF(r→−r←).
La combinazione delle equazioni [7]÷[11] dà un'equazione della curva potenziale-corrente mostrata nella fig. 3, che è interpretabile direttamente come un'espressione della velocità della reazione elettrodica in funzione del potenziale dell'elettrodo. Si tratta di una doppia curva esponenziale; da una parte e dall'altra del punto di equilibrio (j=0) la funzione ha un andamento esponenziale semplice. Questa relazione fu trovata empiricamente nel 1905 da Julius Tafel ed è conosciuta nella sua forma logaritmica come legge di Tafel. L'incremento esponenziale della velocità non può continuare all'infinito, man mano che il potenziale dell'elettrodo si allontana dal valore di equilibrio. Benché la stessa reazione elettrodica sia caratterizzata da una velocità massima, un altro processo incomincia a controllare la velocità prima che sia raggiunto tale limite: il trasporto dei reagenti da e verso l'interfaccia. Esso dipende dalla geometria dell'interfaccia, dalle condizioni idrodinamiche nell'elettrolita e dalla composizione di questo. Se l'elettrodo ha una forma semplice e l'elettrolita è ben mescolato, un semplice modello ideato da Walther Nernst può essere usato per descrivere il trasporto di massa nel caso che il reagente non sia carico, o si trovi in un elettrolita con grande eccesso di ioni non reagenti.
Nernst suppose che la concentrazione del reagente fosse uniforme nell'elettrolita fino a una distanza δ dalla superficie dell'elettrodo e che, entro δ, il trasporto di massa seguisse la legge di diffusione di Fick, secondo cui la velocità di diffusione è proporzionale al gradiente di concentrazione tramite una costante di proporzionalità D, detta coefficiente di diffusione. In effetti, Nernst suppose pure che la concentrazione variasse linearmente dal suo valore alla distanza δ (uguale alla concentrazione nella massa) al suo valore all'interfaccia dell'elettrodo. Perciò, per lo ione Fe2+ nella reazione [6] la velocità di trasporto può essere scritta:
[12] formula

dato che questa velocità per unità di superficie può essere anche espressa come densità di corrente. Nella [12], m indica la concentrazione nella massa. Se la reazione elettrodica è accelerata in direzione anodica variando il potenziale verso valori positivi rispetto al potenziale di equilibrio, vicino all'elettrodo si consumano ioni ferrosi; perciò la concentrazione locale [Fe2+] decresce. Ciò causa un aumento della velocità di diffusione data dalla [12], che va di pari passo con la reazione elettrodica. Tuttavia, quando [Fe2+] diventa molto piccola rispetto a [Fe2+]m, la [12] mostra che si è raggiunta una velocità limite, cioè j raggiunge il valore limite jl.
Elettroanalisi
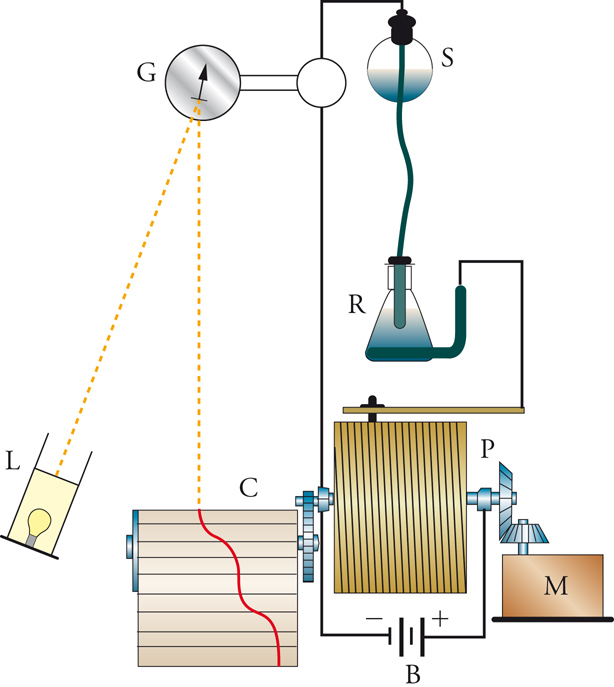
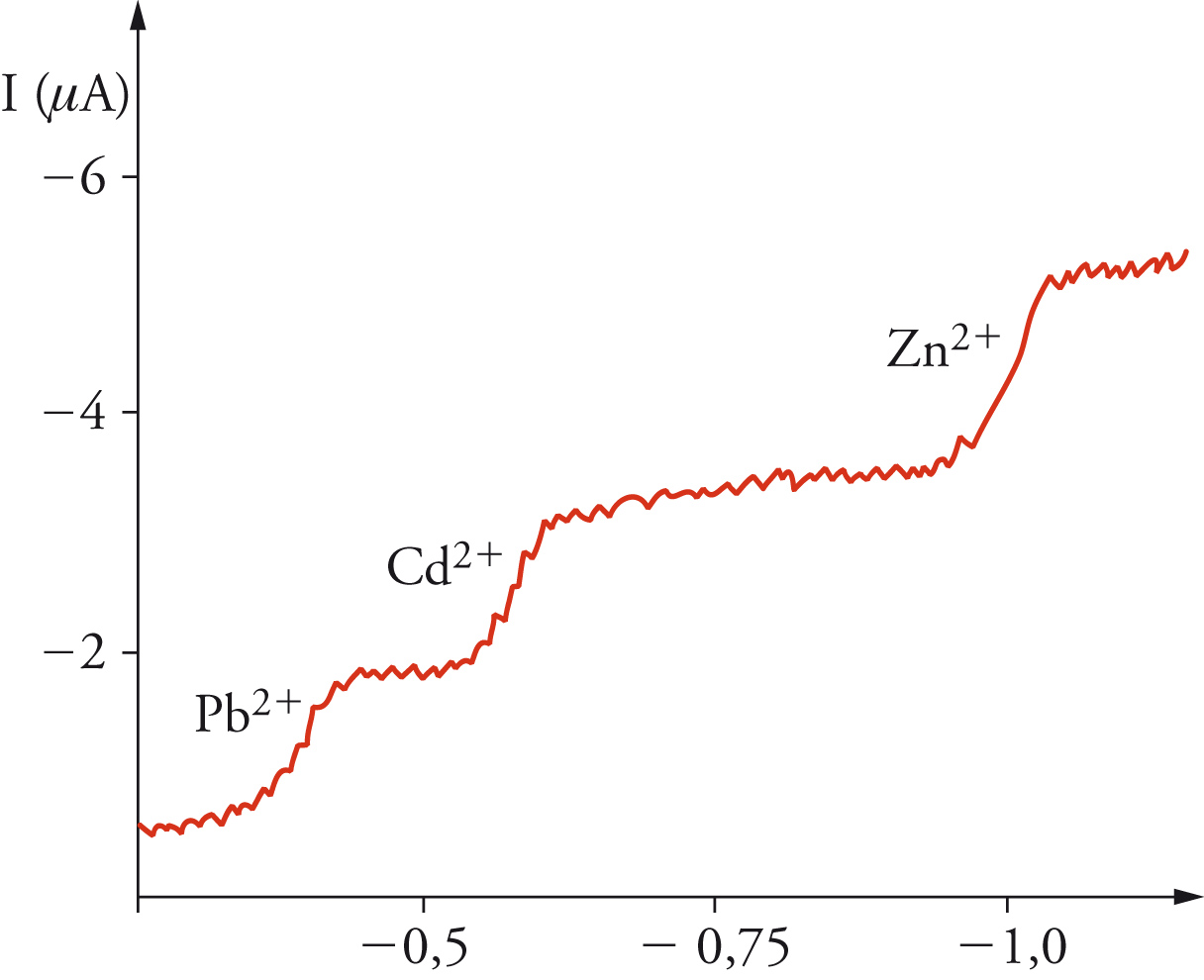
La moderna chimica elettroanalitica ebbe inizio nel 1922 a Praga con l'invenzione del polarografo da parte di Jaroslav Heyrovský, il quale impiegò un elettrodo a goccia di mercurio, che formava una nuova goccia ogni 3÷4 secondi (fig. 4). Ciò forniva una superficie dell'elettrodo pulita e riproducibile e, nello stesso tempo, un ben definito campo di diffusione attorno all'elettrodo. Sebbene le condizioni di quest'ultimo siano più complesse di quelle che conducono all'equazione [12], si ottiene un tipo analogo di corrente limite, anch'essa direttamente proporzionale alla concentrazione della specie reattiva nella massa della soluzione, che quindi essere usata per una rapida analisi quantitativa. Nello stesso tempo, il potenziale al quale avviene la reazione di una data specie è caratteristico della specie stessa, sicché diventa possibile anche un'analisi qualitativa. La caratteristica forma della curva corrente-potenziale è mostrata nella fig. 5. Ogni reazione elettrodica dà origine a una cosiddetta onda polarografica e, in casi favorevoli, è possibile analizzare una miscela in un unico esperimento. L'intervallo di potenziali che si può usare è limitato dalla decomposizione dell'elettrodo stesso (dissoluzione anodica del mercurio) e del solvente (sviluppo catodico di idrogeno). Quest'ultimo processo è molto lento sul mercurio, sicché l'intervallo di potenziale è notevolmente esteso nella direzione negativa. Heyrovský ideò un polarografo registratore automatico (fig. 4), che fu il primo apparecchio analitico automatizzato. Il potenziometro cilindrico che faceva variare il potenziale applicato era fatto girare da un motorino (inizialmente un meccanismo a orologeria), che faceva ruotare simultaneamente un foglio di carta sul quale veniva registrata la corrente. Tutta la curva corrente-potenziale poteva così essere registrata direttamente in circa 4 minuti. Fin verso il 1950, la registrazione veniva fatta fotograficamente, ma poi divennero più comuni i registratori a penna scrivente.
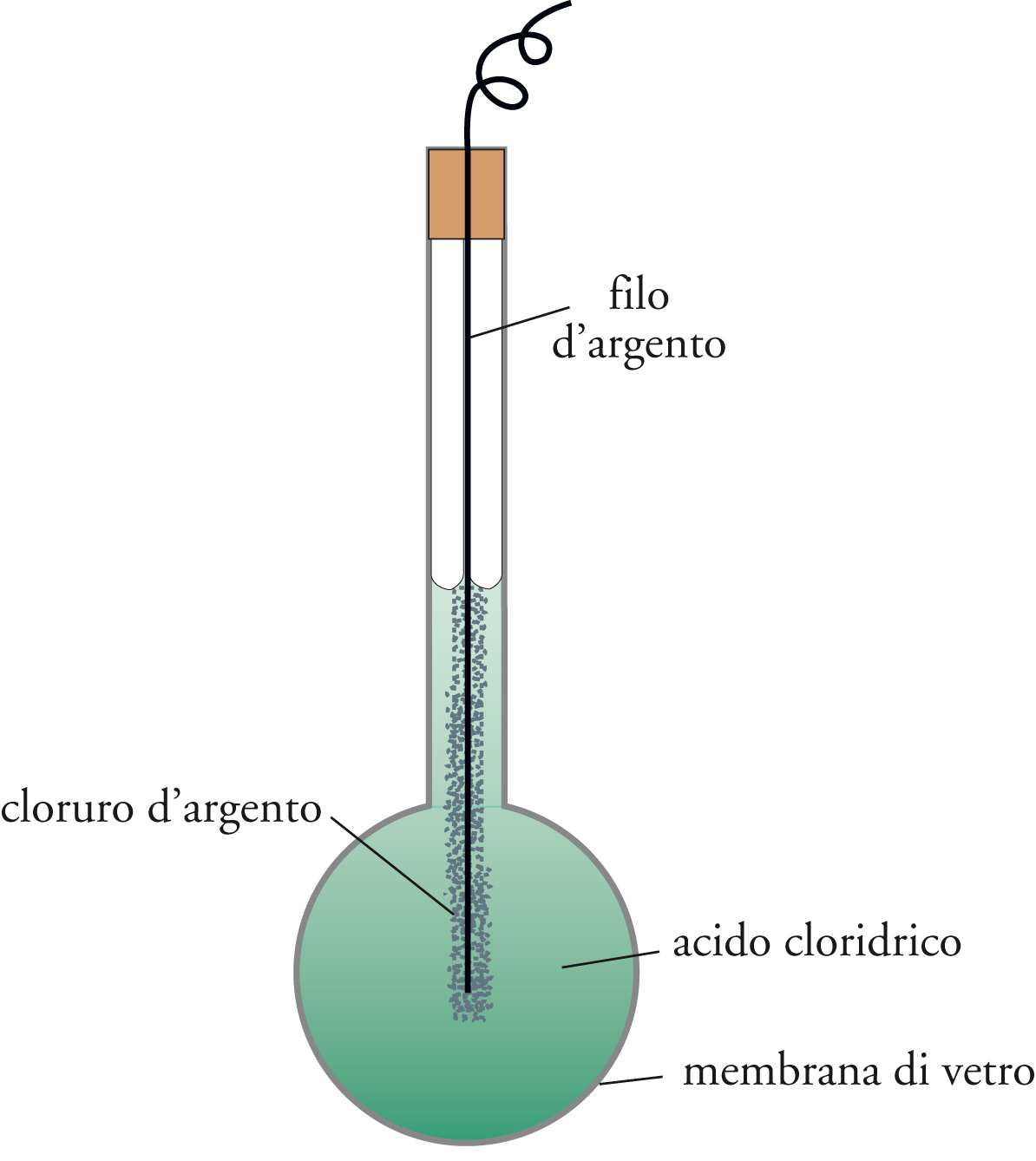
Un tipo del tutto diverso di dispositivo elettroanalitico è basato sulla misura di un potenziale di equilibrio e sull'impiego di un'equazione come la [5] per determinare la concentrazione dello ione con cui l'elettrodo è in equilibrio. Sebbene questa possibilità sia esistita fin dall'inizio del XX sec., essa fu sfruttata ampiamente solo nella forma dell'elettrodo a vetro usato per misurare il pH (=−log[H+]). Questo elettrodo è costituito da un sottile bulbo di vetro, dentro il quale si trovano una soluzione di acido cloridrico e un elettrodo ad argento-cloruro d'argento (fig. 6). Il vetro ha la struttura di un silicato contenente ioni di metalli alcalini che possono essere sostituiti da altri ioni, in particolare da ioni idrogeno provenienti da una soluzione in contatto con l'elettrodo.
Questo processo di scambio, che avviene sulla superficie esterna del vetro, dà origine a una differenza di potenziale (misurabile fra l'elettrodo posto entro il bulbo di vetro e un elettrodo di riferimento a contatto con la soluzione in esame) che dipende logaritmicamente dalla concentrazione idrogenionica, secondo l'equazione di Nernst. Modificando opportunamente la composizione del vetro, l'intervallo di pH a cui l'elettrodo ancora funziona può essere esteso fino a 11 (ambiente notevolmente alcalino), oppure l'elettrodo può essere reso sensibile a uno ione di metallo alcalino. Questi furono i primi elettrodi ionici selettivi o ISE (Ion selective electrodes), ai quali più di recente se ne sono aggiunti molti altri. Il primo elettrodo di questa nuova serie, che diede buoni risultati, è stato quello per gli ioni fluoruro costruito nel 1966, a base di LaF3 solido.
Nel 1967 furono realizzati alcuni pratici elettrodi liquidi a scambio ionico, costituiti da una fase liquida, immiscibile con la soluzione in esame (normalmente acquosa), che conteneva un agente complessante, altamente specifico per lo ione da analizzare e insolubile nella fase acquosa. Ne risulta una distribuzione dello ione in esame (e degli altri ioni), che ubbidisce alle leggi dell'equilibrio di Donnan e che dà luogo a un potenziale di membrana dipendente dalla concentrazione ionica nella soluzione nel modo descritto dall'equazione di Nernst. Il principio basilare è simile a quello dell'elettrodo a vetro, ma il reticolo di silicato solido è sostituito da un agente complessante in soluzione. L'efficienza di questi elettrodi dipende dalla selettività dell'agente complessante e dalla possibilità che hanno altri ioni di attraversare l'interfaccia, in relazione anche alla loro concentrazione nella soluzione. Un recente sviluppo è stato la combinazione di elettrodi selettivi e di dispositivi semiconduttori, i cosiddetti transistori iono-selettivi a effetto di campo o ISFET (Ion sensitive field effect transistor). Questi elettrodi iono-selettivi, benché misurino ioni in soluzione, possono essere adattati ad altri tipi di misure, per esempio all'analisi dei gas, facendo in modo che il gas si sciolga in uno strato sottile di soluzione e produca una variazione rivelabile in una composizione ionica.
Fonti di energia elettrochimica
Le fonti di energia elettrochimica possono essere distinte in celle primarie, celle secondarie e celle a combustibile.
Celle primarie
Dentro una cella primaria si formano dei prodotti chimici, mentre altri vengono consumati, con produzione di elettricità; una volta che i secondi siano esauriti, la cella cessa di funzionare. La maggior parte delle primitive sorgenti di energia elettrochimica, compresa la pila di Volta, erano celle primarie. Quella che ha avuto più successo finora è la cella a zinco/biossido di manganese, inventata da Georges Leclanché nel 1867. La cella originale conteneva una soluzione acquosa di cloruro d'ammonio, ma in seguito l'elettrolita fu sostituito da una pasta e si ottenne la cosiddetta pila a secco. La reazione all'anodo consiste nella dissoluzione di zinco:
[13] Zn → Zn2++2e−
la reazione al catodo è complicata; all'inizio probabilmente essa è:
[14] MnO2+H++e− → MnOOH.
Sono state costruite delle varianti del modello originale: le più comuni sono la pila a elettrolita alcalino e quella con anodo di magnesio; entrambe queste pile hanno una densità di energia più alta di quella della pila originale di Leclanché.
Celle secondarie
Le celle secondarie si comportano come le primarie, salvo che in esse i prodotti chimici, dopo che si sono consumati, possono essere rigenerati in situ facendo passare una corrente in direzione opposta: viene così ripristinato lo stato iniziale della cella. In tal modo, una buona cella secondaria può essere rigenerata più volte. La cella secondaria che ha avuto più successo, inventata da Gaston Planté nel 1859, è la cella a piombo/acido, che è usata come batteria di accensione in tutte le automobili (se ne consumano 108 esemplari all'anno), nonché come principale fonte di energia nella maggioranza dei veicoli elettrici e in parecchi altri sistemi che richiedono accumulo di elettricità. Il funzionamento di questo tipo di cella dipende dall'esistenza di tre stati di ossidazione stabili, tutti presenti come composti solidi relativamente insolubili in acido solforico concentrato. L'elettrodo positivo è costituito da una pasta di biossido di piombo supportata da una griglia di piombo. Durante la scarica, il biossido viene ridotto a solfato di piombo (o a una miscela di questo con PbO):
[15] PbO2+2H++H2SO4+2e−→PbSO4+2H2O.
L'elettrodo negativo è costituito da un'analoga griglia di piombo che supporta una pasta di piombo finemente suddiviso. Durante la scarica il piombo viene ossidato a solfato di piombo:
[16] Pb+H2SO4→PbSO4+2H++2e−.
Quando la cella si è parzialmente o completamente scaricata, è possibile invertire queste reazioni applicando una fonte di elettricità esterna con un voltaggio più alto di quello prodotto dalla cella stessa. In condizioni ottimali, si possono ottenere circa 400 cicli completi, se la scarica è quasi completa a ogni ciclo. Il voltaggio di una cella completamente carica è di 2,2 V. Altri tipi diffusi di celle secondarie sono la cella al nichel/cadmio e quella al nichel/ferro. La loro densità di energia è paragonabile a quella della batteria al piombo/acido (≃25 Wh/kg), ma esse hanno un ciclo vitale molto lungo (diverse migliaia di cicli).
Celle a combustibile
I due tipi di celle esaminati contengono una modesta carica di reagenti, che, ovviamente, limita la quantità di elettricità prodotta prima che la cella debba essere sostituita (cella primaria) o ricaricata (cella secondaria). In una cella a combustibile i reagenti sono forniti continuamente, di modo che la cella funziona in uno stato stazionario, limitato solo dalla stabilità dei suoi componenti. Così, in linea di principio, una cella a combustibile potrebbe sostituire i generatori convenzionali, fondati su una combinazione di combustione/macchina termica/dinamo, e avrebbe una maggiore efficienza, non perché il processo venga completato in un unico stadio, ma perché evita l'impiego di una macchina termica, la cui efficienza ha un limite superiore assoluto espresso dall'efficienza di una macchina termica ideale di Carnot. La prima cella a combustibile fu descritta da William Grove nel 1839. In essa la reazione anodica era l'ossidazione dell'idrogeno:
[17] H2 → 2H++2e−
e la reazione catodica era la riduzione dell'ossigeno:
[18] O2+4H++4e− → 2H2O.
Entrambe le reazioni avvenivano su elettrodi di platino. La cella era quindi la stessa usata per l'elettrolisi dell'acqua, ma con il flusso di corrente in direzione opposta. La reazione complessiva:
[19] 2H2+O2 → 2H2O
è la combustione dell'idrogeno.
Sebbene Grove fosse riuscito a illuminare un'aula per conferenze con primitive lampade a incandescenza alimentate dalla sua cella a combustibile, l'applicazione pratica delle celle aventi come combustibile l'idrogeno si realizzò soltanto nella seconda metà del XX sec. e fu possibile soprattutto in seguito al lavoro pionieristico di Francis T. Bacon. La prima applicazione si ebbe nei veicoli spaziali con equipaggio, dove l'uso di un combustibile leggero e facilmente disponibile, nonché la produzione di acqua, molto utile, fecero di queste celle a combustibile la soluzione ideale per affrontare il problema energetico. Gli elettrodi di Bacon erano di nichel poroso, con pori di due diverse dimensioni, che permettevano un intimo contatto fra gas ed elettrolita senza ingolfamento dell'elettrodo. In alcuni modelli successivi è stato impiegato un foglio poroso di teflon/carbonio, che fa da supporto a un catalizzatore di metallo, spesso costituito da platino finemente suddiviso, per catalizzare le reazioni elettrodiche.
Elettrolisi industriale
Il processo elettrochimico industriale impiegato su più vasta scala è l'elettrolisi di una soluzione di cloruro di sodio per produrre cloro e idrossido di sodio. Il processo di base è semplice: all'anodo si produce cloro secondo la reazione:
[20] 2Cl− → Cl2+2e−
mentre al catodo si producono idrogeno e alcali secondo la reazione
[21] 2H2O+2e− → H2+2OH−.
Una buona produzione di Cl2 e NaOH dipende dall'esclusione della reazione anodica concorrente
[22] 2H2O → O2+4H++4e−
e dalla presenza di un mezzo di separazione dell'alcali dalla salamoia originale.
Questi problemi furono risolti originariamente da Hamilton Y. Castner e Karl Kellner, i quali, nel 1894, elaborarono un processo a due stadi. L'elettrolisi veniva realizzata in una cella con un catodo di mercurio e un anodo di carbone e forniva cloro e amalgama di sodio. Per evitare l'inquinamento da mercurio dell'ambiente, nel 1950 questo processo venne sostituito, soprattutto negli Stati Uniti e in Giappone, con quello a un solo stadio che ha luogo in una cella a diaframma. Questa è divisa in due compartimenti da un diaframma fatto di asbesto impregnato di polimero; in effetti, il diaframma è attaccato al catodo, ragion per cui il compartimento catodico è piccolo e l'NaOH prodotto è estratto attraverso la rete catodica. Il diaframma non è selettivo, sicché parte degli ioni cloro lo attraversa in direzione del catodo e l'alcali è sempre contaminato da NaCl. Per evitare un'eccessiva contaminazione la reazione non è portata fino a un'elevata concentrazione di NaOH, il che rende necessaria una successiva fase di concentrazione. La cella a membrana rappresenta un ulteriore miglioramento, in quanto il diaframma è rimpiazzato da una membrana selettiva per i cationi o addirittura per il sodio: questo permette la produzione di una concentrazione più elevata di NaOH. Le membrane sono polimeri perfluorurati con catene laterali che terminano in gruppi solfonici o carbossilici. Le proprietà di queste membrane impongono un limite alle dimensioni delle unità, ma esse sono in via di rapido sviluppo.
Dopo quello per produrre cloro e alcali, il processo elettrochimico industriale impiegato su più vasta scala è l'estrazione dell'alluminio. Il minerale usato è la bauxite, che dopo la purificazione è costituita essenzialmente di allumina, Al2O3, un ossido che fonde a 2020 °C. La scoperta, fatta nel 1886 da Charles M. Hall e Paul Héroult, che l'allumina si scioglie nella criolite (Na3AlF6), per dare, intorno ai 1000 °C, un fluido conduttore di elettricità, rese possibile la produzione industriale di alluminio su vasta scala. Entrambi gli elettrodi sono di carbonio; il catodo costituisce il fondo della cella, su cui si accumula l'alluminio: in realtà, è quest'ultimo che funziona da catodo effettivo. L'anodo si consuma man mano che reagisce con l'ossigeno e dev'essere ripristinato continuamente. Perciò, la reazione globale della cella è la seguente:
[23] 2Al2O3+3C → 4Al+3CO2.
Altri processi di preparazione di metalli per elettrolisi di sali fusi sono quelli del magnesio, del sodio e del litio. Si è anche cercato di realizzare l'estrazione elettrolitica di metalli come il titanio, ma finora non si è riusciti a mettere a punto un processo in grado di sostituire il vecchio metodo.
Per l'estrazione del rame e dello zinco si usano soluzioni acquose, ma il metodo elettrolitico viene impiegato solo per il 10% della produzione del primo e per il 50% di quella del secondo. Anche quantità più piccole di altri metalli sono prodotte elettroliticamente. Processi simili sono usati per la raffinazione dei metalli rame (Cu), nichel (Ni), cobalto (Co), piombo (Pb), zinco (Zn) in soluzione acquosa e alluminio (Al) allo stato fuso. Anche alcuni composti organici sono prodotti elettroliticamente. Il processo più usato è il metodo Monsanto per l'idrodimerizzazione dell'acrilonitrile ad adiponitrile, un intermedio nella produzione del nylon:
[24] 2CH2=CHCN+2H2O+
+2e−→CN(CH2)4CN+2OH−.
Molte altre ossidazioni e riduzioni organiche sono ottenute per via elettrolitica su piccola scala industriale. In alcune di esse si usa un sistema intermediario inorganico di ossidoriduzione: è quest'ultimo che, in realtà, reagisce all'elettrodo, mentre la reazione desiderata è completata come processo omogeneo. La natura specifica delle reazioni elettrolitiche, insieme al fatto che altri reagenti possono spesso essere esclusi, le rende adatte alla preparazione di prodotti puri; c'è quindi da aspettarsi un'espansione dell'elettrochimica industriale in questa direzione.
Finitura e lavorazione dei metalli
I metodi elettrochimici vengono utilizzati per preparare metalli. Ciò può essere fatto depositando il metallo o rimuovendolo. La deposizione di un oggetto metallico completo è chiamata elettroformatura ed è usata per produrre lamine sottili, reticelle, tubi o bande senza giunzioni, e oggetti più complessi, come le guide d'onda. Gioielli e medaglie d'argento e d'oro sono formati in questo modo, ma la maggior parte dell'elettroformatura è eseguita con nichel. La quantità di metallo depositata nell'elettroplaccatura è di gran lunga minore di quella depositata nell'elettroformatura. Generalmente, l'applicazione di uno strato sottile serve per ottenere proprietà di superficie migliori, senza un grande aumento dei costi. Esempi tipici sono la cromatura (che, generalmente, consiste nella deposizione di uno strato di cromo molto sottile su uno strato di nichel più spesso) e la placcatura in oro di contatti di molti componenti elettronici, di conduttori nelle tavole dei circuiti e di alcuni condensatori. Per assicurare una buona aderenza, l'elettroplaccatura deve essere effettuata su una superficie pulita e questa sovente è ottenuta elettroliticamente. Spesso, il processo comincia con la deposizione di un monostrato di atomi, seguita da nucleazione e crescita degli strati successivi.
Gli aspetti fondamentali di questi processi sono stati molto studiati e la loro cinetica è stata analizzata in termini di stadi elementari. Questo tipo di analisi dev'essere fatto in condizioni accuratamente controllate, che sono piuttosto lontane da quelle di un bagno di placcatura pratico, dove l'obiettivo è generalmente quello di formare sull'oggetto un deposito microcristallino uniforme, spesso rifinito con una lucidatura a specchio. La capacità di un bagno di placcatura di formare un deposito uniforme è chiamata potere ricoprente. Un bagno con buon potere ricoprente darà un deposito uniforme su un oggetto di forma complessa. In linea di principio, una densità di corrente uniforme si ottiene lavorando sulla parte della curva corrente-potenziale (che è quasi piatta), dove la densità di corrente è indipendente dal potenziale, cosicché una distribuzione di potenziale complessa su un oggetto, per esempio a spirale, condurrà a un deposito uniforme.
Sebbene questo risultato possa essere ottenuto lavorando in condizioni limitate dalla diffusione, spesso non si ottiene una buona forma di deposito, ma vengono prodotti dendriti o polveri: un maggior successo si consegue con l'uso preliminare di una reazione chimica, come la dissociazione di un complesso. Ciò promuove anche la dissoluzione dell'anodo, che mantiene costante la composizione del bagno. Frequentemente, si usano complessi cianidrici e allo stesso tempo agenti tensioattivi, allo scopo di modificare le condizioni della deposizione, per adsorbimento sulla superficie. Di solito, questi tensioattivi sono realizzati empiricamente per ottenere lucentezza, livellamento, compensazione degli sforzi nel deposito e per facilitare la rimozione di idrogeno, che, altrimenti, potrebbe formare bolle aderenti alla superficie. Si tratta, generalmente, di molecole organiche associate a gruppi contenenti zolfo, che le rendono capaci di attaccarsi alla superficie dell'elettrodo. In alcuni casi, le leghe possono essere elettroplaccate controllando attentamente le condizioni del potenziale e la composizione del bagno. Oggetti non metallici possono essere placcati dopo essere stati rivestiti di un materiale conduttore, come la grafite.
Il trattamento anodico di un metallo ha come risultato la formazione di una pellicola protettiva. Sottili pellicole di ossido proteggono metalli come l'alluminio e il cromo, il che spiega la loro stabilità nell'uso quotidiano. Comunque, la pellicola sull'alluminio può essere inspessita e le sue proprietà possono essere modificate per fini ornamentali: variando lo spessore, si possono produrre colori d'interferenza o, alternativamente, si possono incorporare nella pellicola, inizialmente porosa, pigmenti che poi vengono fissati chiudendo i pori con un trattamento in acqua bollente. Molti oggetti d'alluminio e di sue leghe sono trattati in questo modo, migliorandone l'aspetto e aumentandone la resistenza alla corrosione.
Corrosione
La corrosione è il processo attraverso cui i metalli reagiscono con sostanze presenti nel loro ambiente per diventare composti, generalmente ossidi, perdendo così le loro proprietà, spesso con risultati catastrofici. L'esempio più noto è l'arrugginimento del ferro. La corrosione determina uno spreco molto rilevante di materiale, che può comportare costi assai considerevoli, fino ad alcuni punti percentuali del prodotto interno lordo di uno stato tecnologicamente avanzato. Sebbene la reazione complessiva possa essere scritta nella forma di una reazione chimica, già Davy si accorse che essa è la somma di due reazioni elettrochimiche, come la reazione in una cella elettrochimica. Davy applicò questo concetto nel 1824, quando fu consultato dall'Ammiragliato britannico a proposito della corrosione delle navi di legno rivestite di rame. Egli mostrò che si poteva impedire la corrosione, attaccando al rame un pezzo di un metallo più ossidabile (ferro, zinco), noto come anodo di sacrificio; esso si scioglie nell'acqua di mare a un potenziale più negativo del rame, così che quest'ultimo mantenga un potenziale a cui è stabile; in tal modo il rame diventa il luogo di una reazione catodica, che normalmente è la riduzione dell'ossigeno sciolto nell'acqua di mare. Questa combinazione di una reazione di corrosione anodica in una certa zona e di una reazione catodica in un'altra, può avvenire anche su scala più piccola, quando due diversi metalli sono a contatto fra loro in un ambiente umido, o anche quando zone distinte di uno stesso metallo si trovano in condizioni diverse, per esempio in differenti stati di tensione o di esposizione all'ossigeno. In generale, questo fenomeno è noto come corrosione per azione locale.
Può anche accadere che la reazione di corrosione e la reazione catodica a essa accoppiata avvengano nella stessa zona di un metallo omogeneo. In condizioni di stato stazionario, si stabilisce un potenziale stazionario noto come potenziale misto. Questo concetto fu introdotto contemporaneamente, nel 1932, da Alexander N. Frumkin e da Louis P. Hammett e Arthur E. Lorch, per spiegare la cinetica di dissoluzione di amalgami di metalli alcalini; fu poi sviluppato, nel 1938, da Carl Wagner e Wilhelm Traud e applicato alle reazioni di corrosione in generale. Le condizioni per la corrosione non dipendono solo dall'attività del metallo e dall'aggressività dell'ambiente, ma anche dalla natura dei prodotti di corrosione: se sono solubili, la dissoluzione del metallo può continuare incontrollata; nel caso in cui, invece, si formi un prodotto insolubile, la situazione è più complessa: se tale prodotto forma uno strato isolante aderente, la corrosione può fermarsi rapidamente. Questa è la ragione della stabilità dell'alluminio e del magnesio in molti ambienti, a dispetto della loro alta reattività. Il ferro, d'altro canto, è meno reattivo, ma l'ossido (ruggine) che si forma è normalmente poroso, conduttore e poco aderente, sebbene in certe condizioni possa accrescersi in una forma aderente come una pellicola sottile, rendendo il ferro passivo.
Questa spiegazione fu data da Faraday e, sebbene molto contestata, essa si è rivelata corretta. Il cromo forma una pellicola passiva migliore e la presenza di una piccola quantità di cromo nel ferro (con nichel) conferisce le sue proprietà protettive all'acciaio inossidabile. L'uso di pellicole protettive passive o di vernice oppure di un altro metallo (rame, zinco), è uno dei modi per ritardare o prevenire la corrosione. Tuttavia, questo sistema può causare problemi se la pellicola è perforata, perché in questo caso la corrosione si concentra in una superficie piccola. Questo e altri tipi di inomogeneità causano corrosione per vaiolatura o corrosione interstiziale. Se, inoltre, il metallo è sotto sforzo, la corrosione può essere accentuata dalla formazione di cricche per corrosione sotto sforzo. L'uso dello zinco nella galvanizzazione è un modo per combattere questi fenomeni: se avviene la perforazione, lo zinco agisce da anodo di sacrificio, mentre il ferro esposto funge da zona catodica.
Si può evitare la corrosione anche modificando il potenziale, o con l'uso di un anodo di sacrificio, oppure utilizzando una fonte di potenziale esterna e un anodo non corrodibile: questo procedimento è noto come protezione catodica. Poiché sul metallo da proteggere può svilupparsi idrogeno, c'è il rischio che questo possa sciogliersi nel metallo facendolo diventare fragile. Un'alternativa è quella di usare un potenziale più positivo, per formare una pellicola passiva anodicamente. Questa protezione anodica, a sua volta, richiede un controllo attento del potenziale, per mantenere la superficie nella regione di potenziale in cui la pellicola passiva è stabile, altrimenti la corrosione può perfino essere accentuata. Le condizioni generali per la corrosione possono essere predette su base termodinamica, in quanto questa fornisce informazioni riguardo gli intervalli di potenziale e di composizione dell'ambiente nei quali il metallo è stabile. In altre regioni può avvenire la corrosione. Queste previsioni furono chiaramente riassunte da Marcel Pourbaix nei diagrammi potenziale-pH, che forniscono un utile punto di partenza per una discussione sulla corrosione, ma, naturalmente, non prendono in considerazione la cinetica delle reazioni; inoltre, le condizioni reali sono generalmente molto più complesse di quelle rappresentate nei diagrammi di Pourbaix. La tecnica dettagliata della corrosione rimane un fatto sperimentale, necessario per una comprensione teorica del fenomeno.
Bioelettrochimica
Molti processi biologici fondamentali possono essere considerati sostanzialmente elettrochimici, poiché implicano trasferimento di carica alle interfacce, sebbene non comportino sempre lo stesso tipo di cambiamento del portatore di carica, tipico delle reazioni discusse sopra. Le interfacce sono membrane, la cui struttura è complessa. Esse consistono, tipicamente, di uno strato bimolecolare di fosfolipidi, in cui le catene idrocarburiche (idrofobe) sono dirette all'interno e i gruppi fosfolipidici (idrofili) formano le due superfici esterne. In questa struttura sono inserite le proteine e altre grandi molecole, che svolgono le funzioni specifiche della membrana. Strutture simili sono state prodotte artificialmente e sono conosciute come membrane lipidiche a doppio strato o BLM (Bilayer lipid membranes). Un doppio strato lipidico puro ha una resistenza molto alta che, però, può essere abbassata addizionando varie sostanze che diminuiscono anche la resistenza delle membrane biologiche.
Sono stati identificati tre meccanismi di trasporto di piccoli ioni inorganici attraverso queste membrane: (a) il moto termico delle molecole può determinare la formazione temporanea di una lacuna di dimensioni molecolari, attraverso cui può passare uno ione. Questi eventi sono rari, sicché la resistenza intrinseca della membrana è alta; (b) il piccolo ione può complessarsi con una specie neutra, solubile nella membrana, passare attraverso quest'ultima e quindi essere rilasciato dal complesso dall'altra parte della membrana. L'agente complessante o trasportatore (carrier) può, in effetti, essere presente solo nella membrana. Un tipico carrier è la molecola macrociclica valinomicina, che si adatta ai cationi alcalini più grossi, ma che non può spostare il rivestimento di idratazione di Li+ o di Na+; (c) un terzo meccanismo è quello delle molecole capaci di formare canali. Ne è un esempio tipico la gramicidina A, un peptide con struttura a elica e con la parte esterna lipofila, per cui è stabile nella membrana. Se l'elica è orientata con il suo asse perpendicolare al piano della membrana, gli ioni possono passare lungo quest'asse e attraversare la membrana.
La gramicidina A, in effetti, permette il trasferimento di protoni preferenzialmente rispetto ad altri ioni monovalenti. Una volta che il trasporto di uno ione sia reso possibile dalla presenza nella membrana di un carrier o di un formatore di canali, esso può essere indotto da un'adatta forza motrice, come un aumento di concentrazione o di potenziale da un lato della membrana rispetto all'altro. Questo è il cosiddetto trasporto passivo. In molte situazioni biologiche, tuttavia, il trasporto sembra avvenire contro un gradiente di concentrazione o di potenziale e viene allora chiamato trasporto attivo. Quest'ultimo è il risultato dell'accoppiamento del trasporto con una reazione metabolica connessa con l'ossidazione di un 'combustibile' biologico, oppure con l'uso di energia immagazzinata in un legame polifosforico dell'adenosintrifosfato (ATP), che viene rotto per formare adenosindifosfato (ADP) e H2PO4−.
Questi processi sono catalizzati da enzimi. L'energia deriva originariamente dall'ossidazione di materiale organico durante la respirazione. Se questa ossidazione coinvolgesse direttamente l'ossigeno molecolare respirato dall'organismo, l'energia sarebbe trasformata in calore e non sarebbe, quindi, disponibile per promuovere altri processi. L'ossidazione, perciò, deve avvenire attraverso una serie di reazioni che sono sempre vicine all'equilibrio. Così l'energia è disponibile per compiere lavoro utile, all'incirca come lo è in una cella elettrochimica. Una serie di reazioni accoppiate di ossidoriduzione, che formano una catena trasportatrice di elettroni, si verifica nella membrana interna di un organulo (una regione specializzata della cellula), chiamato mitocondrio.
Secondo Peter D. Mitchell, l'energia prodotta viene immagazzinata sotto forma di un'aumentata attività dei protoni nei mitocondri. Una serie quasi simile di reazioni avviene nella fotosintesi, ma in questo caso, naturalmente, l'energia iniziale è fornita dalla luce incidente. Questa viene assorbita dalla clorofilla con formazione di una coppia elettrone-lacuna, che viene poi separata, tramite reazione con sistemi adiacenti di ossidoriduzione, in un organulo chiamato tilacoide. Ci sono 2 fotosistemi paralleli ed è richiesto un totale di 4 quanti di luce per ridurre ogni molecola di CO2. Molte delle molecole singole coinvolte in queste catene trasportatrici di elettroni sono state studiate con tecniche elettrochimiche, nel tentativo di capire i dettagli di questi processi estremamente complessi, che sono ancora oggetto di molte discussioni. Dal tempo di Galvani si conosce l'intima relazione fra l'attività neuromuscolare e l'elettricità. Si sospettava che un qualche tipo di conduzione fosse responsabile della propagazione dell'informazione attraverso i nervi. Originariamente si credeva che la velocità di trasmissione degli impulsi nervosi fosse infinita, ma von Helmholtz mostrò, nel 1850, che essa in effetti è di circa 30 ms−1, cioè molto inferiore alla velocità di conduzione elettronica in un metallo: il fenomeno, d'altronde, non può essere spiegato in termini di semplice conduzione ionica, perché in tal caso richiederebbe campi elettrici molto elevati. La spiegazione fu trovata in seguito a esperimenti effettuati verso la metà del XX sec., soprattutto da parte di Alan L. Hodgkin e Andrew F. Huxley, i quali usarono microelettrodi inseriti nelle fibre nervose (assoni) del calamaro gigante, che sono molto grandi. Si trovò che, nello stato di riposo, la composizione all'interno dell'assone era piuttosto differente da quella all'esterno. Cosa particolarmente notevole, la concentrazione degli ioni sodio era 50 mM all'interno e 460 mM all'esterno, mentre quella degli ioni potassio era 400 mM all'interno e 10 mM all'esterno. Inoltre, il potenziale elettrico all'interno della membrana era di 70 mV più negativo di quello esterno.
L'eccitazione di un'estremità della fibra causa un aumento locale della permeabilità della membrana agli ioni Na+ e il potenziale di membrana cambia segno, diventando di circa +50 mV: il cosiddetto potenziale di azione. A questa variazione consegue una propagazione dello stato di eccitazione lungo la membrana, ed è quest'onda di alterazione della permeabilità che conduce il segnale lungo la fibra nervosa, piuttosto che un qualche trasporto fisico di ioni in questa direzione longitudinale. Le proprietà delle membrane furono studiate da Hodgkin, Huxley e loro collaboratori non solo allo stato naturale, ma anche sostituendo il materiale interno con soluzioni saline di composizione nota. Si usarono specie ioniche marcate con traccianti radioattivi per seguire il movimento di ognuna di esse in determinate condizioni elettriche. Fu infilato un filo metallico lungo l'assone e venne controllato il potenziale con una pinza di voltaggio inventata a tale scopo. Questo strumento fu il precursore del potenziostato, usato quasi universalmente nell'elettrochimica attuale. Anche la soglia di eccitabilità fu esaminata usando impulsi di corrente. La spiegazione dell'attività nervosa fu tentata in termini di diffusione di ioni in un campo elettrico costante, usando un modello più adatto a una membrana spessa e tenendo conto della capacità elettrica di questa. Tale spiegazione è stata molto discussa ed elaborata, ma il meccanismo molecolare delle variazioni di permeabilità della membrana resta incerto. Un'ipotesi possibile è che si verifichi l'allontanamento di protoni da specie presenti nella membrana sotto l'azione di un elevato campo elettrico locale, con conseguente variazione della configurazione di alcune proteine.
Bibliografia
Bagockij, Skundin 1980: Bagockij, Vladimir S. - Skundin, Aleksandr M., Chemical power sources, London, Academic Press, 1980.
Bagotsky 2006: Fundamentals of electrochemistry, 2. ed., ed-ited by Vladimir S. Bagotsky, Hoboken (N.Y.), Wiley-Interscience, 2006.
Bard 1985: Standard potentials in aqueous solutions, edited by Allen J. Bard, Roger Parsons, Joseph Jordan, New York, Dekker, 1985.
Bockris 1980-1985: Comprehensive treatise of electrochemistry, edited by John O'Mara Bockris e altri, New York, Plenum, 1980-1985, 8 v.
Bond 1980: Bond, Allan M., Modern polarographic methods in analytical chemistry, New York, Dekker, 1980.
Donnan 1911: Donnan, Frederick G., "Zeitschrift für Elektrochemie", 17, 1911, pp. 571-581.
Dubpernell, Westbrook 1978: Proceedings of the Symposium on selected topics in the history of electrochemistry, edited by George Dubpernell, Jack H. Westbrook, Princeton, Electrochemical Society, 1978.
Galvani 1791: Galvani, Luigi, De viribus electricitatis in motu muscolari commentarius, Bononiae, ex. Typographia Instituti Scientiarum, 1791, pp. 363-418.
Gerischer 1961: Gerischer, Heinz, Semiconductor electrode reactions, in: Advances in electrochemistry and electrochemical engineering, New York-London, Wiley Interscience, 1961-1984; v. I, 1961, pp. 139-232.
Gibbs 1875-1878: Gibbs, Josiah W., On the equilibrium of heterogeneous substances, "Transactions of the Connecticut Academy of Arts and Sciences", 3, 1875-1878, pp. 108, 343.
Gouy 1903, 1906: Gouy, Georges, Sur la fonction électrocapillaire, "Annales de chimie et de physique", 1903, s. 7, 29, pp. 145-241; 1906, s. 8, 8, pp. 291-363; 1906, s. 8, 9, pp. 75-139.
Grahame 1947: Grahame, David C., The electrical double layer and the theory of electrocapillarity, "Chemical review", 41, 1947.
Gray 1963: Gray, W.F.M., Shocking discovery, "Journal of the Electrochemical Society", 110, 1963, pp. 210C-211C.
Grotthuss 1806: Grotthuss, C.J. Theodor, Sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'électricité galvanique, "Annales de chimie et de physique", 58, 1806, pp. 54-73.
Hamman 2007: Hamman, Carl H. - Hamnett, Andrew - Vielstich, Wolf, Electrochemistry, 2. ed., Weinheim, Wiley - VCH, 2007.
Hodgkin, Huxley 1945: Hodgkin, Alan L. - Huxley, Andrew F., Resting and action potentials in single nerve fibres, "Journal of physiology", 104, 1945, pp. 176-195.
Izutsu 2002: Izutsu, Kosuke, Electrochemistry in nonaqueous solutions, Weinheim, Wiley - VCH, 2002.
Koryta 1982: Koryta, Jiri, Ions, electrodes and membranes, Chichester-New York, Wiley, 1982.
Levic 1962: Levic, Ven'jamin G., Physicochemical hydrodynamics, 2. ed., Englewood Cliffs (N.J.), Prentice-Hall, 1962.
Macdonald 1977: Macdonald, Digby D., Transient techniques in electrochemistry, New York, Plenum, 1977.
Mitchell 1968: Mitchell, Peter, Chemiosmotic coupling and energy transduction, Bodmin, Glynn Research, 1968.
Moore 1977: Moore, W.J., Electrochemistry of nerves, in: Special topics in electrochemistry, edited by Peter A. Rock, Amsterdam, Elsevier, 1977, pp. 128-160.
Nernst 1904: Nernst, Walther H., Theorie der Reaktionsgeschwindigkeit in heterogenen Systemen, ‟Zeitschrift für physikalische Chemie", 47, 1904, pp. 52-55.
Parsons 1954: Parsons, Roger, Equilibrium properties of electrified interphases, in: Modern aspects of electrochemistry, edited by John O'Mara Bockris, London, Butterworths, 1954, pp. 103-179.
Pletcher 1982: Pletcher, Derek, Industrial electrochemistry, London, Chapman & Hall, 1982.
Pourbaix 1966: Pourbaix, Marcel, Atlas of electrochemical equilibria in aqueous solutions, Oxford, Pergamon, 1966.
Rowlinson, Widom 1982: Rowlinson, John S. - Widom, Benjamin, Molecular theory of capillarity, Oxford, Clarendon, 1982.
Tafel 1905: Tafel, Julius, Über die Polarisation bei kathodischer Wasserstoffentwicklung, ‟Zeitschrift für physikalische Chemie", 50, 1905, pp. 641-712.
Volta 1800: Volta, Alessandro, On the electricity excited by the mere contact of conducting substances of different kinds, "Philosophical transactions of the Royal Society of London", 90, 1800, pp. 403-431.
Wagner, Traud 1938: Wagner, Carl - Traud, Wilhelm, Über die Deutung von Korrosionsvorgängen durch Überlagerung von elektrochemischen Teilvorgängen und über die Potentialbildung an Mischelektroden, ‟Zeitschrift für Elektrochemie und angewandte physikalische Chemie", 44, 1938, pp. 391-402.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


