Enzima
Enciclopedia on line
Sostanza di natura proteica (una volta detta fermento) che ha proprietà di accelerare una reazione chimica specifica senza esser consumata e senza entrare nei prodotti finali della reazione. Le sostanze sulla cui trasformazione gli e. agiscono sono dette substrati. A differenza dei catalizzatori inorganici, molti e. sono capaci di agire solo su un determinato tipo di substrato, specifico per ognuno di essi.
Cenni storici
Nella storia dell’enzimologia si possono distinguere tre grandi periodi. Il primo va dal 17° sec. alla fine del 18° e comprende le prime osservazioni scientifiche sulla fermentazione alcolica, sulla putrefazione e su certe trasformazioni chimiche, come la digestione della carne operata dal succo gastrico, molto più tardi riconosciute di natura enzimatica. Tra gli scienziati più insigni che si occuparono di questi problemi vanno ricordati J.B. van Helmont, T. Willis, G.E. Stahl, J.J. Becher, A. van Leeuwenhoeck nel 17° sec. e H. Cavendish, L. Spallanzani e A.-L. Lavoisier nel 18° secolo. Quest’ultimo per primo dimostrò (1789) che la fermentazione alcolica era un processo che seguiva le leggi quantitative della chimica e da allora ebbe inizio il periodo moderno dell’enzimologia.
Nel secondo periodo, che occupa tutto il 19° sec., si estesero e approfondirono le conoscenze sulle fermentazioni in generale e su quella alcolica in particolare, e inoltre furono scoperte e studiate alcune trasformazioni chimiche enzimatiche negli animali e nelle piante. Ricordiamo il contributo di A.-P. Dubrunfaut, che preparò (1830) un estratto di malto che idrolizzava l’amido in maltosio, e quelli di P.-J. Robiquet e A.-F. Boutron (1830), e contemporaneamente di Chalard, che nelle mandorle amare individuarono una sostanza, più tardi denominata emulsina, capace di scindere l’amigdalina. Nel 1831 E.F. Leuchs descrisse l’azione della ptialina salivare sull’amido; nel 1833 A. Payen e J.-F. Persoz isolarono l’amilasi dall’orzo tallito e la purificarono; nel 1836 T. Schwann scoprì la pepsina nel succo gastrico; nel 1837, indipendentemente, C. Cagniard De la Tour, Schwann e F. Kutzing provarono che il lievito, prima creduto un composto chimico, era invece un organismo vivente; nel 1856 L. Corvisart scoprì la tripsina. Tra il 1860 e il 1875 L. Pasteur dimostrò che, perché avvenissero la fermentazione alcolica e le altre fermentazioni, era necessaria la presenza di microrganismi vivi. In conseguenza fu fatta una distinzione fra fermenti organizzati, indissolubilmente legati a cellule vive e non estraibili da queste in forma attiva (per es., il lievito, i batteri della fermentazione lattica, acetica ecc.), e fermenti non organizzati, secreti da cellule (per es., pepsina del succo gastrico) o estraibili dalle cellule in forma attiva (es. l’amilasi dell’orzo in germinazione); questi ultimi furono denominati e. da W. Kühne nel 1878.
Nel 1897 E. Buchner provò che dal lievito si poteva spremere un succo capace di compiere la fermentazione alcolica, dimostrando che la distinzione tra fermenti organizzati e non organizzati non aveva più ragione di essere; da allora ebbe inizio il periodo attuale della enzimologia. In seguito, le conquiste più importanti sono state: la definizione dei fattori che condizionano le reazioni enzimatiche; la scoperta della somiglianza tra la fermentazione alcolica del lievito e la glicolisi nelle cellule animali; la scoperta dei coenzimi e della partecipazione delle vitamine alla loro costituzione; la purificazione del primo e. in forma cristallina e il riconoscimento della sua natura proteica (J.B. Sumner 1926) e quindi la cristallizzazione di numerosi di essi; la scoperta della sintesi enzimatica di polisaccaridi come l’amido, il glicogeno ecc.; la scoperta che certi processi patologici sono determinati da alterazioni di funzioni enzimatiche; la identificazione di meccanismi di immagazzinamento e di trasferimento dell’energia negli organismi.
Struttura degli enzimi
Gli e. possono essere costituiti da proteine semplici (per es., ribonucleasi) che determinano da sole sia la specificità sia la reazione, oppure da proteine coniugate. In questo caso nell’e. è presente anche un gruppo non proteico che prende il nome di gruppo prostetico quando è saldamente legato al resto della molecola; quando invece la parte proteica e quella non proteica sono facilmente dissociabili, la prima prende il nome di apoenzima, la seconda quello di coenzima; dall’unione delle due parti si ottiene l’e. completo o oloenzima, che viene semplicemente chiamato enzima. Quando l’e. è costituito da proteine coniugate, partecipano alla reazione il gruppo prostetico o il coenzima che fungono da trasportatori di elettroni o di gruppi chimici, mentre la porzione proteica o l’apoenzima determinano la specificità. L’attività di alcuni e. richiede pertanto la presenza di molecole non proteiche (coenzimi o anche ioni metallici) che vengono detti cofattori.
La specificità, che differenzia i biocatalizzatori dai catalizzatori inorganici, presenta vari tipi e gradi: 1) specificità di legame: l’e. attacca solo un determinato legame chimico; 2) specificità di gruppo chimico: l’e. attacca solo un determinato gruppo chimico; 3) specificità di substrato: l’e. attacca solo certi composti e non altri che siano anche suscettibili di subire la stessa reazione. Alcuni e. sono specifici per gli isomeri geometrici o per quelli cis-trans; per es., la fumarasi addiziona acqua a livello del doppio legame dell’acido fumarico (trans) ma è completamente inattiva sull’isomero cis, l’acido malonico.
Per poter funzionare, ogni e. deve formare con il proprio substrato un complesso chiamato substrato-e. (S-E), e poiché la maggior parte dei substrati hanno molecole piccole rispetto a quelle del biocatalizzatore, solo una piccola parte della molecola enzimatica (detta sito attivo) è impegnata nella formazione del complesso substrato-enzima. Il sito attivo è formato da una sequenza di amminoacidi specifica per ciascun e., situata in una regione caratteristica e protetta per ogni molecola enzimatica; la conformazione e la configurazione spaziale del sito attivo permettono che esso si combini solo con un determinato tipo di struttura; l’importanza della struttura tridimensionale è documentata dalle gravi alterazioni dell’attività enzimatica che subentrano quando essa è modificata (per es., dal calore).
Modalità d’azione
Il complesso substrato-e. ha vita molto breve, durante la quale il substrato subisce una modificazione specifica con rottura o formazione di legami covalenti; la formazione del complesso S-E permette che avvengano rapidamente reazioni fondamentali per l’essere vivente, quali la trasformazione e l’utilizzazione degli alimenti (carboidrati, lipidi, protidi ecc.); gli e. pertanto non modificano l’equilibrio di una reazione reversibile, ma possono solo accelerarla in entrambi i sensi. Infatti in natura le reazioni chimiche spontanee sono esoergoniche (in esse cioè vi è una diminuzione di energia libera) e a esse prendono parte solo molecole attivate, le quali posseggono una carica di energia superiore a quella delle molecole normali; questa differenza di energia viene detta energia di attivazione ed è variabile da un sistema all’altro.
Il numero di molecole del sistema che raggiunge lo stato di attivazione; è tanto più piccolo quanto maggiore è la differenza (dislivello) tra l’energia allo stato iniziale e quella allo stato di attivazione; è quindi necessario che diminuisca il dislivello perché un numero elevato di molecole raggiunga lo stato di attivazione e la reazione abbia luogo. La diminuzione del dislivello si può ottenere attraverso due vie: a) innalzando l’energia dello stato iniziale; b) abbassando l’energia di attivazione iniziale. È con questa seconda modalità che agiscono gli enzimi, attraverso la formazione del complesso substrato-e. che richiede un’energia di attivazione minore di quella necessaria al decorso diretto della reazione in assenza di biocatalizzatore. Il substrato unito all’e. subisce le modificazioni proprie della reazione, poi il complesso si scinde, l’e. torna libero e capace di combinarsi con un’altra molecola di substrato per riprendere il ciclo, e il substrato trasformato è rilasciato come prodotto della reazione.
La cinetica delle reazioni catalizzate da e. (tranne la scissione dell’acqua ossigenata da parte della catalasi, per la quale non si conosce la saturazione da substrato) è caratterizzata dal fenomeno della saturazione da parte del substrato: a basse concentrazioni di substrato la velocità di reazione è proporzionale alla concentrazione di esso, ma se si aumenta la concentrazione del substrato, la velocità di reazione tende a crescere in misura minore e non è più proporzionale alla concentrazione di substrato; aumentando ulteriormente la concentrazione, la velocità di reazione diventa costante e indipendente dalla concentrazione del substrato. A questo punto l’e. è saturato (cioè completamente legato) dal substrato; allo stato di saturazione il fattore che limita la velocità della reazione è rappresentato unicamente dalla concentrazione dell’enzima. L’effetto della saturazione portò L. Michaelis e M.L. Menten a formulare, nel 1913, una teoria generale dell’azione e della cinetica enzimatica, che più tardi fu ampliata da G. E. Briggs e J.B.S. Haldane.
L’attività di un e. dipende anche da numerosi altri fattori, quali la temperatura, il cui aumento accelera ulteriormente la velocità di reazione, ma che oltre un certo limite provoca la denaturazione della proteina enzimatica, il pH dell’ambiente in cui si svolge la reazione (il pH ottimale della maggior parte delle reazioni è compreso tra 5 e 8), l’effetto di particolari ioni (Na+, Mn2+, Mg2+), necessari per alcune reazioni.
Regolazione
Negli organismi viventi esistono molteplici meccanismi per la regolazione dell’attività degli e. a seconda delle necessità: tra di essi ricordiamo il fenomeno dell’allosteria, l’inibizione dell’attività enzimatica e l’inibizione o l’attivazione della sintesi enzimatica. Gli e. allosterici sono formati da varie subunità identiche costituite da una o più catene polipeptidiche (monomeri). Ogni subunità possiede un sito attivo e una zona di regolazione che comprende un sito di legame diverso da quello catalitico, a cui si lega una determinata sostanza detta attivatore o effettore allosterico positivo e un sito di legame diverso sia da quello catalitico sia da quello dell’attivatore a cui si lega un’altra sostanza detta inibitore o effettore allosterico negativo. Generalmente, quando gli e. sono scissi in subunità, sono cataliticamente inattivi. L’e. esiste in due forme, una catalitica (R o relax), in cui può combinarsi sia con il substrato sia con l’attivatore, e una inibita (T o tense), in cui può legarsi solo all’inibitore; le due forme sono tra loro in equilibrio, che è influenzato dalle concentrazioni relative del substrato, dell’attivatore e dell’inibitore. Il substrato e gli effettori, fissandosi all’e., spostano l’equilibrio verso la forma R o T, e in particolare l’attivatore (che in alcuni casi può essere rappresentato dal substrato stesso), unendosi all’e., favorisce la forma R che lega il substrato e lo trasforma, mentre l’inibitore (che in alcuni casi può essere rappresentato dal prodotto della reazione) favorisce la forma T che non lega il substrato ed è quindi inattiva. Concludendo, l’attività degli e. allosterici, in particolare quelli implicati nella regolazione del metabolismo e detti perciò e. regolatori, è regolata dalla concentrazione dei vari substrati e dei corrispondenti effettori allosterici.
L’inibizione dell’attività catalitica è detta irreversibile quando implica la modificazione permanente di uno o più gruppi funzionali, prevalentemente al sito attivo, dell’e. e quindi il blocco dell’attività enzimatica (alcune delle sostanze che provocano modificazioni irreversibili sono veleni nervini, alcuni insetticidi ecc.). L’inibizione reversibile, in cui il blocco dell’attività enzimatica può essere rimosso, è detta competitiva quando una sostanza (detta inibitore) simile al substrato usuale, compete con questo per il sito attivo dell’e., impedendo quindi la formazione del complesso substrato-enzima; l’inibizione può essere rimossa aumentando adeguatamente la concentrazione del substrato. L’inibizione reversibile non competitiva avviene quando l’inibitore si lega all’e. o al complesso substrato-e. in una zona diversa dal sito attivo; questo tipo di inibizione non dipende quindi dalla concentrazione del substrato, ma può essere rimosso eliminando gli inibitori: per es., alcuni agenti possono legarsi agli ioni metallici necessari all’attività di certi enzimi.
Proprietà
Numerosissimi sono gli e. isolati e purificati. Molti di essi sono stati anche cristallizzati e ne è stata determinata la struttura primaria (sequenza degli amminoacidi) della molecola proteica. In diversi casi è stata studiata la struttura conformazionale, è stato localizzato il sito attivo ed è stato interpretato il meccanismo d’azione dell’attività enzimatica. La maggior parte degli e. sono situati all’interno delle cellule, nel citoplasma o nelle strutture cellulari ( e. intracellulari); alcuni vengono prodotti dalle cellule, ma sono da queste secreti e agiscono al di fuori di esse ( e. extracellulari), come per es. gli e. del tratto digerente. Questi ultimi vengono generalmente secreti come precursori inattivi ( zimogeni) e a loro volta attivati da altri enzimi. Per trasformare uno zimogeno in e. attivo, occorre rimuovere un peptide che funge da inibitore. Per es., per convertire il tripsinogeno in tripsina è necessario che si stacchi dalla molecola un esapeptide costituito da valina, quattro molecole di acido aspartico e lisina; ciò provoca una modificazione nella configurazione della proteina originaria, che diventa attiva.
Essendo di natura proteica, gli e. presentano tutte le proprietà delle proteine; sono denaturati, e quindi inattivati, dal calore e da altri agenti denaturanti chimici e fisici. Le recenti acquisizioni sulle relazioni intercorrenti tra geni ed e. e sulla sintesi enzimatica hanno permesso di interpretare alcuni difetti genetici come l’espressione di alterazioni a carico del DNA. Queste alterazioni possono provocare sia il blocco della sintesi di una proteina enzimatica ( enzimopenia) sia la formazione di una proteina difettosa con perdita o modificazione dell’attività catalitica: ne derivano blocchi metabolici che danno luogo a quadri morbosi di diversa natura.
Molti e. trovano impiego terapeutico: per uso topico (come nel caso della ialuronidasi), per via orale (insufficienze digestive, sindromi da malassorbimento ecc.) e parenterale (impiego endovenoso di e. fibrinolitici nel trattamento precoce dell’infarto del miocardio e di sindromi tromboemboliche ecc.).
Classificazione
L’Unione internazionale di biochimica distingue gli e. in sei gruppi (classi), sulla base del tipo di reazione che catalizzano: ossidoreduttasi, transferasi, idrolasi, liasi, isomerasi, ligasi o sintetasi. Ogni classe è stata contraddistinta da un numero, rispettivamente da 1 a 6. Ciascuna di queste classi è stata suddivisa in sottoclassi: gli e. di ogni sottoclasse sono stati raggruppati in sotto-sottoclassi. Ogni sottoclasse e sotto-sottoclasse è stata contraddistinta da un numero. Infine un altro numero è stato assegnato a ogni specifico enzima. Secondo questa classificazione ufficiale ciascun e. viene a essere individuato da una serie di 4 numeri (la lattico deidrogenasi è indicata con i numeri 1.1.1.27).
Applicazioni industriali
Molti e. sono utilizzati in svariati processi industriali: per la preparazione di prodotti alimentari (formaggi, birra, vino, pane ecc.), nell’industria dei detergenti, nell’industria del cuoio, nell’industria tessile ecc. Si hanno e. estratti da organismi sia vegetali sia animali; la maggior parte, però, di quelli usati è di origine microbica. Fra gli e. estratti da vegetali va ricordata soprattutto la papaina, usata nella stabilizzazione della birra, nell’ammorbidimento delle pelli ecc. Fra gli e. di origine animale sono da citare: la pancreatina, usata nella sbozzimatura dei tessuti, nell’industria del cuoio ecc.; la pepsina, usata per preparare idrolizzati proteici, per idrolizzare la gelatina dei film e delle lastre fotografiche onde consentire il ricupero dell’argento ecc.; la rennina, usata per coagulare il latte nella produzione del formaggio.
Gli e. di origine microbica sono quelli di maggiore interesse industriale in quanto la loro disponibilità non viene a essere legata a quella degli organismi animali o vegetali, ma solo alla capacità produttiva dei microrganismi che nella maggior parte dei casi può essere espansa a piacere. Fra questi sono da ricordare: a) gli e. proteolitici (proteasi), che si possono suddividere in base all’origine (proteasi batteriche, da funghi ecc.) e al campo di pH entro cui esplicano la loro attività in modo ottimale; b) gli e. capaci di agire sui glicidi: particolarmente importanti sono le amilasi del malto, in grado di scindere l’amido, la glicosoisomerasi, che ha la proprietà di isomerizzare il glucosio a fruttosio, le pectinasi, che si utilizzano per eliminare le pectine (o sostanze analoghe) dai succhi di frutta.
Gli e. hanno assunto grande importanza anche in chimica analitica: la loro specificità consente, infatti, di determinare una sostanza senza eseguire le lunghe e costose operazioni di separazione, in quanto essa viene selettivamente trasformata dall’e. in un composto di facile rivelabilità (per es., tramite una colorazione). Nell’ambito di tali applicazioni, particolarmente interessanti sono gli elettrodi a e., sostanzialmente elettrodi indicatori di specie molto comuni (CO2, NH3), che nella membrana che li costituisce contengono un e. immobilizzato chimicamente o fisicamente, sicché è possibile determinare, a seconda dell’e. immobilizzato, tutte quelle sostanze che per reazione enzimatica forniscono la specie rispetto alla quale l’elettrodo è indicatore (per es., un amminoacido in presenza di particolari e. fornisce ammoniaca, che può essere rivelata dall’opportuno elettrodo).
Per quanto concerne la produzione degli e. per via microbica, le tecniche seguite per lo sviluppo dei vari microrganismi necessari alla produzione di tali e. sono quelle tipiche dei processi di fermentazione, cioè mediante colture sia in superficie che sommerse.
E. di restrizione
Le proprietà di alcuni e. di azione specifica per molecole polinucleotidiche quali DNA o RNA sono alla base delle manipolazioni cromosomiche dell’ingegneria genetica. Tali e. vengono comunemente definiti e. di restrizione: appartenenti alle endonucleasi, riconoscono una specifica sequenza di DNA e producono due tagli, uno in ciascun filamento, generando delle estremità 3′OH e 5′P. La scoperta degli e. di restrizione nei batteri, la comprensione delle loro modalità di azione e la loro utilizzazione per analizzare la struttura del gene hanno rivoluzionato gli studi di ;genetica dal 1970 in poi. Proprio per questi studi a W. Arber, H.O. Smith e D. Nathans è stato conferito il premio Nobel per la fisiologia o la medicina nel 1978.
Gli e. di restrizione presenti nei batteri proteggono le cellule dalle infezioni virali riconoscendo il DNA estraneo e degradandolo. Vengono più propriamente chiamati e. di restrizione-modificazione in quanto sono in grado sia di frammentare il DNA estraneo, restringendo il numero di fagi che possono infettare la cellula, sia di modificare, mediante metilazione, il DNA del batterio in modo tale che l’e. non possa riconoscere e digerire il DNA batterico stesso. In alcuni casi tuttavia il DNA virale è metilato (batteriofagi T2, T4, T6); si accresce così la resistenza virale all’azione delle endonucleasi di restrizione batteriche e può avvenire (ciclo litico) l’infezione fagica.
Tutti gli e. di restrizione fanno tagli su due filamenti singoli di DNA, un taglio per filamento. Le disposizioni dei tagli sono diverse: o si verificano nel centro di simmetria della sequenza e si generano così frammenti con estremità smusse (blunt ends), o sono situati in modo asimmetrico rispetto alla linea di simmetria e si generano così estremità adesive (sticky end). Dato che gli e. di restrizione riconoscono una sequenza unica e specifica, ognuno di essi genera una famiglia unica di frammenti da una particolare molecola di DNA, anche se vi sono alcuni e. detti isoschizomeri, che hanno la stessa specificità e generano famiglie identiche. La famiglia di frammenti di DNA di differente lunghezza, ma con estremità identiche, generata da un singolo e. è evidenziata dall’elettroforesi su gel di agarosio; i frammenti si separano su gel perché migrano a una velocità che è in funzione del loro peso molecolare. Confrontando fra di loro le dimensioni dei frammenti di DNA in seguito al trattamento con una combinazione di vari e. di restrizione, si può determinare l’ordine relativo delle sequenze di riconoscimento (siti di restrizione) presenti nella molecola e stimare anche il numero di nucleotidi presenti fra un sito e il successivo. Si ottiene in tal modo un diverso tipo di mappa cromosomica chiamata mappa di restrizione. Dal confronto di mappe siffatte per due o più geni correlati è possibile stimare l’eventuale omologia che li lega. Per es., le mappe di restrizione che codificano l’emoglobina sono rimaste in larga misura inalterate nell’uomo, nell’orango e nello scimpanzé per tutto il periodo intervenuto dopo che queste specie iniziarono a divergere 5-10 milioni di anni fa.
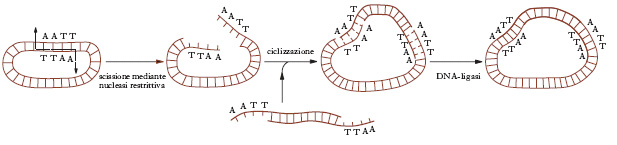
Gli e. di restrizione hanno grande importanza negli esperimenti di ingegneria genetica perché permettono la saldatura di due frammenti di DNA qualsiasi, purché generati dalla medesima endonucleasi restrittiva e dotati quindi di estremità complementari. Nel caso di e. che producono blunt ends non coesive è possibile aggiungere sequenze di nucleotidi per creare le estremità coesive necessarie al legame (linkers). Quando le due estremità si sono congiunte mediante l’accoppiamento delle basi complementari, la saldatura definitiva avviene a opera della DNA-ligasi, un e. che provvede a formare i legami fosfodiesterici fra le estremità opposte di ciascun filamento di DNA. Mediante l’impiego di e. di restrizione e DNA-ligasi si può inserire un segmento qualunque di DNA in elementi capaci di autoduplicazione, quali i plasmidi o i batteriofagi che costituiscono i cosiddetti veicoli di clonaggio dei geni (v. fig.). Le molecole ibride di DNA così ottenute vengono reintrodotte nei batteri resi temporaneamente permeabili alle macromolecole. Mentre i batteri proliferano si replica anche il plasmide, producendo un numero enorme di copie del frammento originario del DNA. Con queste metodiche è stato possibile clonare geni umani che codificano per l’insulina o l’ormone della crescita e far produrre pertanto queste sostanze in grande quantità da ceppi batterici opportunamente selezionati.
Spesso si ottiene il DNA da clonare scindendo l’intero genoma di una cellula mediante un’endonucleasi restrittiva specifica. Dal genoma di mammifero si ottengono una grande quantità di frammenti (da 105 a 107) e il processo di clonazione, di conseguenza, dà luogo a milioni di colonie differenti, ciascuna delle quali ospita un plasmide in cui è inserita una diversa sequenza di DNA genomico. Si deve poi selezionare la rara colonia il cui plasmide contenga la regione di DNA genomico che interessa e la si deve fare proliferare fino a formare una popolazione di cellule numerose o clone. La tecnica cui normalmente si ricorre per selezionare il clone desiderato si basa sull’impiego, in funzione di sonda, di acidi nucleici marcati con isotopi radioattivi complementari al DNA clonato. Mediante questo metodo si è resa possibile la localizzazione ( mappatura) di un gran numero di geni sui cromosomi umani.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


