Sistema reticoloendoteliale
Enciclopedia del Novecento (1982)
Sistema reticoloendoteliale
di Angelo Baserga e Giuseppe Castaldi
SOMMARIO: 1. Antiche e moderne vedute sul sistema reticoloendoteliale (SRE). □ 2. Anatomia e istologia del sistema reticoloendoteliale: a) i primi criteri di classificazione; b) concezioni attuali. □ 3. Origine e cinetica delle cellule del sistema reticoloendoteliale: a) monociti; b) macrofagi. □ 4. Ruolo fisiologico del sistema reticoloendoteliale: a) fagocitosi; b) funzione antixenica; c) funzione citocateretica; d) funzione emopoietica; e) funzione trofica; f) funzione metabolica. □ 5. La patologia del sistema reticoloendoteliale: a) premesse; b) le classificazioni tradizionali; c) le nuove classificazioni proposte; d) malattie del SRE con differenziazione cellulare scarsa o variabile; e) malattie del SRE con differenziazione cellulare moderata; f) malattie caratterizzate dalla proliferazione di istiociti ben differenziati. □ 6. Appendice: hairy cell leukemia o reticoloendoteliosi leucemica. □ Bibliografia.
1. Antiche e moderne vedute sul sistema reticoloendoteliale (SRE).
Il termine ‛sistema reticoloendoteliale' fu usato per la prima volta nel 1913 da L. Aschoff e dai suoi collaboratori per indicare un complesso di elementi cellulari aventi in comune la capacità di inglobare nel citoplasma alcuni coloranti o metalli introdotti nell'organismo vivente sotto forma di sospensioni colloidali. Non erano mancati precursori di questa teoria; ma fu solo dopo gli studi della scuola di Aschoff (Kiyono, Landau, ecc.) che si considerò come un'unità funzionale, o SRE, l'insieme delle cellule mesenchimali dotate di attitudine fagocitaria (da ϕάγω = mangio) o granulopessica, capaci cioè di ingerire materiali particolati. Questo concetto, all'origine rigidamente aderente a criteri di ordine morfofunzionale, fu poi esteso alla patologia: i tentativi di formulare una teoria unitaria delle malattie del SRE presentarono però il difetto, comune a tutte le teorie unitarie, di una eccessiva generalizzazione, con conseguente inclusione nel nuovo edificio nosografico di una quantità enorme di quadri morbosi eterogenei, sicché il concetto di SRE ne risultò modificato, ampliato e talvolta svisato.
Gli studi di questi ultimi decenni hanno permesso di riportare il concetto di SRE entro i confini originali segnati da Aschoff, seppure con qualche ulteriore limitazione, sulla base della miglior conoscenza morfologica, funzionale e cinetica degli elementi cellulari che lo costituiscono. Inoltre, sulla scia del rinnovato interesse per i problemi del linfocito e dell'immunità, si sono enormemente accresciute le conoscenze sull'attitudine fagocitaria delle cellule del SRE, sulla loro capacità di distinguere il proprio (self) dall'estraneo (not self) e sul loro intervento nei fenomeni immunitari. Il grande progresso degli studi di fisiopatologia del SRE ha permesso, infine, di giungere a una migliore delimitazione della sua patologia: delimitazione che, seppur forse non ancora fissata in modo decisivo, può ora costituire la base per un inquadramento nosologico più completo.

Tenuto conto di queste premesse, la storia degli studi sul SRE potrebbe essere suddivisa in sei fasi: 1)1881-1913, la fase anteriore ad Aschoff, cioè la fase della raccolta dei primi dati sull'attività fagocitaria di cellule mesenchimali. In questo periodo Mečnikov chiamò macrofagi (cioè grandi mangiatori) le cellule in grado di ingerire particelle di grandi dimensioni, come eritrociti, spermatozoi, protozoi, ecc., in contrapposizione ai leucociti polimorfonucleati, che chiamò microfagi, in quanto capaci di ingerire solo particelle di dimensioni minori (come ad esempio i Batteri). Mečnikov ebbe anche il merito di riconoscere le strette relazioni esistenti tra le cellule fagocitarie della milza, dei linfonodi, del midollo osseo e del tessuto connettivo, e coniò per questi elementi il termine unitario di ‛sistema macrofagico'; 2) 1913-1924, la fase dell'identificazione del SRE dal punto di vista morfofunzionale. Questo periodo va dal primo lavoro di Landau e McNee, del 1913, alla monografia riassuntiva di Aschoff del 1924. La delimitazione morfologica del SRE fu resa possibile dall'impiego dei cosiddetti ‛coloranti vitali', come il rosso neutro, il blu pirrolo e il blu tripan, sostanze che vengono fagocitate come tali dai macrofagi, consentendone quindi la colorazione e il riconoscimento nell'organismo vivente Nella tab. I sono indicati i vari tipi di cellule appartenenti, secondo Aschoff, al SRE: esse vennero classificate e suddivise sulla base della maggiore o minore capacità fagocitaria; 3) 1924-1932, la fase delle prime descrizioni di affezioni del reticoloendotelio (per esempio, l'osservazione di una reticolosi aleucemica da parte di Letterer) e delle prime pubblicazioni isolate su argomenti di patologia reticoloendoteliale, tra le quali possiamo citare le iniziali classificazioni della patologia del SRE a opera di Schittenhelm, di Epstein e di Zoia. Nel frattempo si estendevano gli studi di istologia e di fisiologia del SRE, e molti autori (v., per esempio, Volterra, 1927) proponevano la sostituzione del nome e del concetto di sistema reticoloendoteliale con quello di sistema reticoloistiocitario (SRI); 4) 1932-1946, la fase dell'elaborazione sistematica delle conoscenze sulle malattie del SRE autonomamente intese, con la pubblicazione delle ampie trattazioni riassuntive di Baserga e di Pittaluga, fino alla fondamentale monografia di Cazal (v., 1946); 5) 1957-1969, la fase del rinnovato interesse per la fisiopatologia del SRE anche da parte degli autori americani, che precedentemente (nonostante i bei lavori di Jaffè e altri) avevano alquanto sottovalutato le concezioni di Aschoff. Si ebbe in quegli anni un rifiorire di studi, fino alla costituzione di una società apposita (The Reticulo-Endothelial Society) e di riviste specializzate (come il ‟Journal of RES"). Le ricerche di tale periodo servirono a meglio precisare molti aspetti della morfologia, delle funzioni e della cinetica delle cellule fagocitanti in condizioni normali e patologiche; 6) dal 1969 a oggi, la fase della sostituzione del concetto di SRE con quello di MPS: le nuove conoscenze sulle cellule fagocitanti degli anni sessanta portarono alla formulazione del concetto di ‛sistema dei fagociti mononucleati' (MPS, Mononuclear Phagocyte System) a opera di un gruppo di studiosi riunitisi a Leiden nel 1969 (Langevoort, Cohn, Hirsch, Humphrey, Spector e van Furth). Secondo la loro proposta il concetto di MPS dovrebbe sostituire quello di SRE, in quanto presenta il vantaggio della semplicità e della comprensione del comportamento dei fagociti mononucleati in condizioni fisiologiche e patologiche. Successive riunioni del gruppo originario e di numerosi altri ricercatori (v. van Furth, 1975 e 1980) hanno confermato l'applicabilità del concetto di MPS ai problemi immunologici, anatomopatologici e clinici. La stessa World Health Organization (WHO) ha accettato, fin dal 1972, una versione lievemente modificata di questo concetto.
2. Anatomia e istologia del sistema reticoloendoteliale.
a) I primi criteri di classificazione.
La prima delimitazione anatomica del sistema reticoloendoteliale fu quella di Aschoff, che assunse il carattere dell'attività granulopessica quale criterio di attribuzione al SRE di elementi cellulari diversi. Per Aschoff appartengono al SRE quelle cellule che accumulano in forma granulare le sostanze coloranti acide; di tali elementi era tuttavia noto il diverso comportamento per quanto riguarda la facilità, la rapidità e l'intensità di colorazione e la grandezza dei granuli accumulati. Su queste differenze dell'attività granulopessica si basò uno speciale raggruppamento proposto dallo stesso Aschoff. Egli operò una prima distinzione fra elementi a spiccata attività granulopessica, costituenti nel loro insieme il SRE in senso lato, ed elementi a debole attività granulopessica, cioè i fibroblasti e gli endoteli dei vasi sanguigni e linfatici, non identificabili come appartenenti al SRE. Egli inoltre distinse un primo gruppo di cellule, cellule reticoloendoteliali propriamente dette o SRE in senso stretto (cui appartengono le cellule del reticolo della polpa splenica, dei nodi della corteccia e dei cordoni midollari dei linfonodi e del tessuto linfatico in genere, i reticoloendoteli dei seni dei linfonodi, dei seni sanguigni della milza, dei capillari epatici, dei capillari del midollo osseo, della corteccia surrenale, dell'ipofisi), e un secondo gruppo al quale appartengono gli istiociti (elementi mobili dei tessuti connettivi), gli splenociti, gli endotelioleucociti, gli istiociti ematici.
Secondo la concezione di Aschoff, gli istiociti potevano derivare dagli elementi del primo gruppo, cioè del SRE in senso stretto, per cui questi ultimi erano anche chiamati istioblasti (Kiyono); d'altra parte, Aschoff ammetteva l'esistenza di cellule che per la finezza dei granuli di materiale colorante assunti, per la delicata rete cromatinica del nucleo, per il grosso corpo cellulare provvisto di prolungamenti sembrano collocabili in posizione intermedia tra gli istiociti e i fibrociti e potrebbero quindi rappresentare una fase intermedia di trasformazione dai primi ai secondi. In seguito prevalse la tendenza a considerare entro limiti più ampi di quelli che erano stati originariamente fissati da Aschoff le caratteristiche anatomiche di identificazione del SRE (Ferrata, Di Guglielmo, Volterra, Pittaluga, Bianchi, Bufano).
Il concetto di cellule reticolari, dibattuto e controverso, si basava essenzialmente sull'osservazione in microscopia ottica di cellule caratterizzate da estese ramificazioni dei prolungamenti citoplasmatici che, se associate, danno l'impressione di una rete tridimensionale. Ma contro la reale unità citoontogenetica di queste cellule erano state sollevate molte obiezioni tra gli anni cinquanta e sessanta. Anzitutto, il reperto morfologico di formazione reticolare può essere ascritto a cellule di varia natura: macrofagi, fibroblasti, endoteli dei seni, cellule interdigitanti e dendritiche varie (v. van Heyden, 1978). Alcuni autori consigliavano semplicisticamente di considerare come cellule del reticolo del midollo osseo le cellule stromatiche che rimanevano dopo l'allontanamento di tutti gli elementi parenchimali, e proponevano criteri analoghi anche per identificare le cellule reticolari dei linfonodi, del fegato, del timo, ecc. Altri preferivano per le cellule del reticolo, e in particolare per le cosiddette cellule reticolari istiocitarie, un inquadramento non ontogenetico, ma funzionale. Già nel 1958 Gall affermava addirittura che la cellula reticolare appare come un mito e che mantenere questo nome non servirebbe ad alcuno scopo utile.
b) Concezioni attuali.
Le attuali conoscenze, fondate sulla funzione - oltre che sulla morfologia e sull'origine - delle cellule mononucleate fagocitanti, hanno permesso di restringere l'eccessivamente vasto sistema ai tipi cellulari riportati nella tab. II. Il loro insieme costituisce, secondo Langevoort e altri (1970), il MPS, cui appartengono anche il monoblasto e il promonocito, cellule midollari che danno origine a ogni elemento del sistema (v. van Furth, 1975).

La morfologia di questi fagociti mononucleati dipende, almeno in parte, dall'organo o dal tessuto in cui hanno sede. La caratteristica morfologica più specifica sarebbe rappresentata da un certo corrugamento della membrana citoplasmatica, riconoscibile in microscopia a contrasto di fase o in microscopia elettronica a scansione. Ma già l'osservazione di strisci colorati con May-Grünwald-Giemsa permette per lo più il riconoscimento degli elementi del sistema nei tessuti emopoietici; ulteriori possibilità di caratterizzazione di queste cellule sono offerte dalla citochimica e dalla microscopia elettronica.
Conformemente alla descrizione che ne danno Bessis (v., 1972), Meuret (v., 1974), Goud e altri (v., 1975) e Lessin e altri (v., 19772), in base alle caratteristiche morfologiche sono identificabili tre principali tipi cellulari del SRE (v. cellula: Fisiologia).
1. Monoblasti e promonociti. Come abbiamo ricordato, sono i precursori midollari dei fagociti mononucleati; i primi, non ancora identificati morfologicamente nell'uomo, si presenterebbero nel topo come elementi rotondeggianti, di diametro compreso fra 10 e 12 μm, con superficie dotata di piccole propaggini e di aspetto lievemente arruffato. Il citoplasma è scarso, fortemente basofilo, agranulato e dotato di qualche vacuolo; la reazione per le esterasi non specifiche risulterebbe positiva nella massima parte degli elementi. Il nucleo, tondeggiante, sarebbe contraddistinto da cromatina nucleare finemente dispersa e dalla presenza di nucleoli. I promonociti si presenterebbero come elementi lievemente allungati, con diametro compreso fra 13 e 34 μm, con citoplasma ancora basofilo, ma contenente fini granuli oltre a qualche vacuolo; in contrasto di fase mostrerebbero, oltre ai granuli e a strutture vescicolari, caratteristiche estroflessioni digitiformi. Le indagini citochimiche darebbero reazioni positive per perossidasi, fosfatasi acida, arilsolfatasi, e soprattutto per le esterasi non specifiche. Il nucleo si presenta voluminoso, reniforme, con indentature profonde; questi aspetti morfologici consentirebbero di distinguere il promonocito dal promielocito. In microscopia elettronica si osserverebbero un apparato di Golgi ben sviluppato, numerosi poliribosomi, scarsi e piccoli granuli elettrondensi, vacuoli e rare membrane ergastoplasmatiche.
2. Monocito. È una cellula del diametro di 12-15 μm, con citoplasma abbondante, grigio-blu, provvisto di fini granulazioni azzurrofile, a volte molto numerose; il nucleo è voluminoso, centrale o più spesso periferico, il più delle volte irregolare, reniforme, con cromatina chiara, a fini filamenti, senza rilevanti aspetti compatti. Non si osservano nucleoli (v. fig. 1). Dal punto di vista citochimico il monocito dimostra una positività per la reazione del paS in forma sia diffusa sia di fini granuli, un'occasionale positività per il sudan nero e per la perossidasi, positività per la citocromossidasi, per le deidrogenasi, per le esterasi non specifiche (reazione inibita dal fluoruro di sodio), per la fosfatasi acida, per l'arilsolfatasi e per la β-glucuronidasi; notevole appare l'attività muramidasica, mentre la reazione per la fosfatasi alcalina risulterebbe negativa. In microscopia elettronica il nucleo si presenta provvisto di uno o due nucleoli circondati da cromatina nucleolo-associata; il citoplasma mostra la presenza di molti ribosomi e poliribosomi, di una scarsa quantità di reticolo endoplasmatico rugoso, di un corpo di Golgi ben sviluppato sito nell'indentatura del nucleo, di numerosi microtubuli e microfibrille, di lisosomi primari (corrispondenti alle granulazioni azzurrofile) e, dopo fagocitosi, di lisosomi secondari. Alla superficie cellulare possono inoltre essere osservate numerose microvillosità e vescicole di micropinocitosi.
3. Macrofagi tessutali. Sono grandi cellule del diametro di 30-40 μm, il cui voluminoso nucleo ovalare o reniforme, con cromatina a maglie regolari, è provvisto di uno o, raramente, più nucleoli. Il citoplasma è assai abbondante, grigio chiaro con zone rosee e bluastre sparse, provvisto di granulazioni azzurrofile; vi si può riscontrare la presenza di vacuoli incolori, a volte assai numerosi, e di ogni tipo di frammenti fagocitati. Le reazioni citochimiche sono analoghe a quelle dei monociti e dipendono, almeno in parte, dal materiale fagocitato. L'esame al microscopio elettronico evidenzia numerosi ribosomi, voluminosi corpi di Golgi, lisosomi primari, reticolo liscio e rugoso, corpi multivescicolari, talvolta numerose fibrille in vario atteggiamento, aspetti di pinocitosi e fagocitosi con presenza di lisosomi secondari.
L'attribuzione di tutte le cellule del SRE a uno di questi tre tipi cellulari non è ovviamente semplice, dal momento che gli elementi mononucleati fagocitanti dell'organismo possono presentare aspetti assai diversi a seconda dell'ambiente che li circonda e del loro stato funzionale: così, nel liquido peritoneale o nei liquidi di altre sierose, essi assumono aspetto rotondeggiante simil-linfocitario (v. fig. 2), mentre nei tessuti si adattano alle fibre collagene e alle cellule circostanti. Negli spostamenti acquisiscono in genere aspetto assai allungato e, durante la pinocitosi, lasciano protrudere lembi citoplasmatici; dopo la fagocitosi possono assumere gli aspetti più diversi, a seconda del tipo e della quantità del materiale inglobato (v. figg. 3 e 4). In certi organi e tessuti e in talune situazioni patologiche, infine, possono presentarsi con aspetti intermedi o addirittura come cellule che nulla sembrano avere in comune con gli elementi mononucleati fagocitanti (come ad esempio gli osteoclasti, v. fig. 5, e le cellule giganti polinucleate delle finestre cutanee alla Rebuck).
Oltre alle cellule descritte, tutta una serie di elementi che con esse non hanno nulla in comune, quanto a origine e natura, si sono dimostrati capaci di fagocitosi facoltativa. Una rassegna di Rabinovitch (v., 1970) ne riporta un grande numero (l'epitelio vaginale, gli epatociti, l'epitelio renale, l'epitelio intestinale, l'epidermide, le mastcellule ecc.) e ne afferma l'importanza per la comprensione generale dei processi fagocitari. Anche se le loro proprietà si sono rivelate assai utili nel chiarimento della funzione dei macrofagi, non ci sembra che queste cellule possano essere inquadrate nel MPS, dal momento che la loro attitudine fagocitaria è facoltativa e modesta, la loro specializzazione funzionale assai sviluppata e la loro origine diversa da quella propria del sistema. Analogamente sono stati esclusi dal MPS diversi tipi cellulari, capaci bensì di fagocitosi, o almeno di micropinocitosi, ma la cui origine è chiaramente mesenchimale, e non emopoietica come quella del MPS. Questi elementi mesenchimali (cellule reticolari, cellule dendritiche, fibroblasti e cellule endoteliali) possono ormai essere distinti dai macrofagi sulla base di tutta una serie di criteri morfologici, di test funzionali in vitro e di peculiari proprietà in vivo.
3. Origine e cinetica delle cellule del sistema reticoloendoteliale.
a) Monociti.
La sede della monocitopoiesi, che ha sempre rappresentato un dibattuto problema, fu inizialmente identificata nel midollo osseo e successivamente nei linfonodi e nel tessuto endoteliale (reticoloendoteliale). Mentre molti ematologi dei decenni passati erano orientati a considerare i monociti come derivanti dagli elementi istiocitari del SRE, la concezione attuale è diametralmente opposta, in quanto considera i monociti quali precursori - non già derivati - delle cellule istioidi dei tessuti; le ricerche recenti, d'altronde, hanno fornito dimostrazioni sempre più convincenti a sostegno del concetto classico della sede midollare della monocitopoiesi.
Il problema è stato affrontato, negli ultimi vent'anni, impiegando la tecnica dei trapianti o delle trasfusioni di varie popolazioni cellulari, marcate con timidina triziata o con altre molecole radioattive, in animali da esperimento. I risultati di questi esperimenti hanno consentito di stabilire con sicurezza nel midollo osseo l'ubicazione degli elementi precursori dei monociti del sangue periferico, cosicché il SRE può essere considerato oggi come un continuum di cellule progressivamente più mature, dai progenitori midollari (monoblasti e promonociti) alla fase circolante monocitaria, quindi ai macrofagi tessutali immaturi e maturi. A monte dei monoblasti sarebbe ancora identificabile un elemento progenitore, capace di dare origine, oltre che alle cellule del SRE, anche a quelle della serie granuloblastica.
Attualmente, per i progressi registrati da questi studi, appare possibile una valutazione globale della monocitopoiesi e della monocitocinetica. Soprattutto grazie al notevole contributo di Meuret (v., 1974) si ammette oggi l'esistenza di un pool midollare di 584 × 106 precursori per kg di peso corporeo, una produzione media di 6,8 × 106 monociti per ora per kg di peso corporeo, un tempo di sintesi del DNA di 9,5 ore e un tempo di generazione media di 12,8 ore. A seguito delle sue ricerche, condotte anche con timidina triziata e con 3HDFP, Meuret è giunto alla conclusione che il ciclo cellulare dei promonociti dura in media 29 ore, così che il tempo disponibile per i precursori durante il loro passaggio attraverso il compartimento di proliferazione è sufficiente allo svolgimento di due cicli generativi successivi. Ciò porterebbe a concludere che la monocitopoiesi è identificabile solamente in un compartimento intramidollare di proliferazione: non vi sono dimostrazioni, infatti, dell'esistenza di uno speciale compartimento di maturazione o di riserva. Il compartimento di proliferazione consta di un sistema di più generazioni di promonociti legati tra loro a modo di catena.
I singoli promonociti percorrono in media due cicli generativi, con un ciclo cellulare medio di circa 29 ore. Il tempo normale dalla cellula staminale fino al passaggio nel sangue circolante sarebbe quindi complessivamente di circa 58 ore. In condizioni normali la capacità di proliferazione dei promonociti viene solo parzialmente utilizzata; in caso di aumentato bisogno di monociti, come ad esempio nelle infezioni e nelle infiammazioni, la riserva di proliferazione viene adeguatamente mobilizzata. I promonociti sembrano essere, in qualsiasi momento del loro passaggio nel compartimento di proliferazione, in grado di migrare dal midollo al sangue circolante: tuttavia il livello di probabilità di questo passaggio è basso nelle cellule giovani e diviene più elevato nelle cellule progressivamente più mature.
Inoltre, sulla base dei risultati di esperimenti di ritrasfusioni di autosangue con monociti marcati, Meuret e Hoffmann (v., 1973) avevano stabilito che il semitempo medio di persistenza in circolo dei monociti è di 8,4 ore, e che nei soggetti sani il rapporto tra il pool dei monociti ‛marginati' e quello dei monociti effettivamente circolanti è di 3,5 : 1. L'elaborazione dei dati consente di ottenere un valore di ricambio medio di 7,49 × 106 monociti/h/kg, il che corrisponde abbastanza bene al valore di 6,8 × 106 monociti/h/kg attribuito alla produzione da parte del midollo. Non è lecito trarre conclusioni rigorose da dati così poco precisi; questi, tuttavia, accettati integralmente consentirebbero di ritenere che la quota di quelle cellule di origine non midollare classificate, sulla base dell'aspetto morfologico, come monociti non può comunque superare il 10% di tutte le cellule valutate come monociti.
Secondo Whitelaw e Batho (v., 1975) il tempo di dimezzamento dei monociti circolanti sarebbe stato sottostimato da Meuret e Hoffmann per motivi tecnici: infatti, la marcatura in vitro dei monociti e la loro ritrasfusione provocherebbero seri danni e quindi una riduzione della sopravvivenza. Sulla base di studi condotti in vivo, Whitelaw e Batho avrebbero dimostrato che i monociti umani rimangono in circolo dalle 36 alle 104 ore, quindi molto più a lungo di quanto rilevato da Meuret e Hoffmann.
Circa le modalità della fuoruscita dei monociti dal circolo vi è ormai generale accordo: tali elementi migrano nel compartimento extravascolare per effetto del caso e non come conseguenza di un processo di senescenza (v. van Furth e Cohn, 1968). Questa migrazione avverrebbe per diapedesi, dopo adesione agli endoteli, soprattutto a livello delle venule postcapillari. Una volta che abbiano raggiunto i tessuti, i monociti vanno incontro a tutta una serie di modificazioni anatomofunzionali, acquisendo in primis una grande capacità fagocitaria e dando origine in definitiva ai macrofagi. Il numero di questi ultimi sarebbe molto superiore a quello dei monociti ematici (400 : 1; v. Osgood, 1954). La morte dei monociti avverrebbe, del tutto accidentalmente, durante la loro permanenza in circolo.
b) Macrofagi.
Abbiamo ormai numerose prove dell'origine monocitaria dei macrofagi tessutali: già le ricerche di Florey (v., 1962) avevano permesso di stabilire la provenienza dai monociti dei macrofagi del tessuto connettivo e delle cavità sierose, e ulteriori studi ontogenetici avevano dimostrato che i fagociti mononucleati non sono presenti nei tessuti finché questi non siano vascolarizzati (v. Andersen e Matthiesen, 1966). Nell'embrione qualche cellula difficilmente classificabile, ma già contrassegnata da alcune caratteristiche dei monociti, si può trovare dalla quarta alla quinta settimana di vita intrauterina. Secondo Kelemen e altri (v., 1979), monociti chiaramente identificabili appaiono nel sangue del cordone ombelicale e del cuore dalla undicesima alla quattordicesima settimana, e costituiscono successivamente dall'1 al 4% di tutte le cellule nucleate; istiociti e macrofagi possono osservarsi negli strisci di sangue solo fino alla quattordicesima settimana. Successivamente è stata dimostrata con estrema chiarezza la derivazione dai monociti del sangue periferico anche delle cellule di Kupffer e dei macrofagi cerebrali e degli alveoli polmonari. Della stessa origine sono risultati i macrofagi liberi della milza, dei linfonodi e del midollo osseo, mentre poco si sa ancora sull'origine dei macrofagi fissi siti negli stessi organi, mancando per ora la prova diretta della loro probabile derivazione monocitaria: il fatto però che le cellule di Kupffer del fegato, classico esempio di macrofagi fissi, derivino dai monociti ematici deporrebbe per un'analoga origine di ogni macrofago fisso.
Un problema non ancora completamente risolto concerne la possibilità o meno che i macrofagi dei tessuti ricircolino attraverso il sangue o la linfa per raggiungere altri distretti dell'organismo. La constatazione che i macrofagi peritoneali marcati migrano verso linfonodi lontani - verso la milza, il fegato e il tessuto sottocutaneo - e che, se iniettati per via endovenosa, possono allogarsi nel fegato, nella milza e nella cavità peritoneale, suggerisce che i macrofagi liberi siano in grado di entrare in circolo e di raggiungere altri organi (v. fig. 6). Per contro, nulla ancora si sa sulle possibilità di mobilizzazione e di migrazione dei macrofagi fissi dei tessuti (v. Roser, 1965; v. Whitelaw e Batho, 1975).
La durata di vita dei macrofagi è lunga: i bassissimi valori di turnover osservati nei macrofagi tessutali (v. van Furth, The origin ..., 1970), e confermati dalla stabilità della distribuzione della popolazione macrofagica marcata nel fegato e nella milza (v. Roser, 1970), lasciano supporre che la sopravvivenza totale dei macrofagi tessutali sia dell'ordine di mesi. D'altra parte è assai difficile osservare queste cellule in corso di mitosi, il che fa pensare a un periodo intercinetico assai prolungato; solo in condizioni particolari, ad esempio dopo intensi stimoli fagocitari o immunitari, l'indice mitotico e quello di marcatura con timidina triziata potrebbero aumentare sensibilmente (v. van Furth, Origin..., 1970; v. Shima e altri, 1972; v. Whitelaw e Batho, 1975). Secondo Cline e Sumner (v., 1972) questi elementi ancora capaci di proliferazione apparterrebbero al gruppo dei macrofagi A immaturi che, a differenza dei macrofagi B maturi, manterrebbero la possibilità di un sia pure limitato divenire cariocinetico (v. fig. 7). A questo proposito merita di essere segnalato il concetto, prospettato da Spector e Mariano, della differenziazione fra granulomi a high turnover - nei quali, per la persistente presenza di un agente macrofagotossico altamente attivo, i macrofagi vengono rapidamente distrutti e sostituiti mediante il reclutamento di nuovi macrofagi - e granulomi a low turnover, indotti da agenti macrofagotossici meno attivi. Quando il focolaio infiammatorio persiste più di una settimana, i macrofagi più vecchi, se non vengono distrutti, tendono a modificarsi nel corso dell'attivazione in cellule epitelioidi e a fondersi in cellule giganti (v. Spector e Mariano, 1975). In definitiva, la cinetica dei macrofagi è complessa e difficilmente valutabile, perché tali cellule posseggono un proprio potenziale di proliferazione (ben studiato, per esempio, da North: v., 1969), la cui grandezza pare largamente autonoma e suscettibile di essere modulata anche all'infuori dell'approvvigionamento di nuove cellule da parte del comparto monocitopoietico midollare.
La morte dei macrofagi avrebbe luogo a livello del polmone e del lume intestinale (v. Bertalanffy, 1964; v. Humphrey, 1970); si dovrebbe inoltre ammettere anche la morte di un buon numero di macrofagi, eventualmente fagocitati da altri macrofagi, per giustificare la costanza del numero dei fagociti mononucleati del sangue in condizioni fisiologiche.
4. Ruolo fisiologico del sistema reticoloendoteliale.
a) Fagocitosi.
L'esame dettagliato di tutte le attività del SRE appare di estrema difficoltà, dal momento che nell'organismo esso interviene in numerosissimi eventi biochimici e fisiologici e che non si può escludere che gli studi futuri ne dimostrino la partecipazione a funzioni che finora non gli sono state attribuite.
La rassegna dei dati offertici dalla letteratura ci permette attualmente di attribuire al SRE le seguenti funzioni generali (v. Bessis, 1972): 1) funzione antixenica; 2) funzione citocateretica; 3) funzione emopoietica; 4) funzione trofica; 5) funzione metabolica.
Poiché la massima parte di queste funzioni si esplica attraverso i meccanismi della fagocitosi e della pinocitosi, è opportuno anzitutto prendere in considerazione la grande capacità fagocitaria che le cellule del SRE mostrano nei confronti di materiali endogeni ed esogeni. Nella terminologia di North (v., 1970), per endocitosi si intende il processo mediante il quale una cellula ingerisce materiale extracellulare, racchiudendolo in invaginazioni della sua membrana citoplasmatica (v. cellula: Fisiologia). Il termine convenzionalmente comprende sia la pinocitosi sia la fagocitosi, espressioni originariamente usate per descrivere l'ingestione cellulare di gocciole lipidiche e, rispettivamente, di particelle solide. L'azione del fagocita sulla particella che deve ingerire può essere distinta in tre fasi: 1) migrazione del fagocita verso la particella; 2) fagocitosi propriamente detta, che si suddivide in a) aderenza della particella al fagocita e b) ingestione della particella; 3) digestione (o trasporto) della particella all'interno del fagocita.
La migrazione del fagocita verso la particella, o viceversa, e la sua adesione a questa sono dovute a meccanismi d'ordine chimico-fisico, quali forze elettrostatiche, forze esistenti fra superfici idrofobiche e idrofiliche, forze di van der Waals, legami covalenti ecc. (v. Jones, 1975), oppure a meccanismi decisamente immunitari. I meccanismi non immunitari (tipico quello che regola la fagocitosi di particelle inerti quale, per esempio, il carbone) operano anche in elementi non monocito-macrofagici, purché capaci di fagocitosi facoltativa nel senso già prospettato (v. Rabinovitch, 1970), mentre quelli immunitari sono caratteristici del SRE. In quest'ultimo caso, perché abbiano luogo la migrazione del fagocita verso la particella, o viceversa, e la fagocitosi propriamente detta, è necessario l'intervento delle opsonine, fattori serici identificati con gli anticorpi (immunoglobuline IgM e IgG) e con il complemento (componenti C1, C4, C2 e C3, reagenti nell'ordine), che agirebbero, in cooperazione o meno (v. Opsonins, 1970; v. Jones, 1975), fissandosi all'antigene; la frazione C3 del complemento, per esempio, acquisterebbe attività proteolitica (precisamente peptidasica), che si manifesterebbe su di un sito recettore specifico della cellula fagocitaria, provocando quindi l'aderenza tra cellula e particella e la conseguente fagocitosi (v. immunologia e immunopatologia: Immunologia generale e Malattie autoimmuni). Circa il meccanismo con cui una cellula capace di fagocitosi verrebbe messa in grado di reagire esercitando la fagocitosi stessa nei confronti di una particella opsonizzata, poco sappiamo: è stata avanzata l'ipotesi che la cellula riconosca un mutamento conformazionale dell'opsonina, che si verifica quando quest'ultima si unisce all'antigene. Pertanto, come osserva North (v., 1970), nell'ambito del sistema fagocitante degli organismi superiori il primo riconoscimento dell'estraneità competerebbe al sistema umorale.
L'attività fagocitaria comporta il dispendio di una discreta quantità di energia, ottenuta attraverso la stimolazione delle vie glicolitiche aerobia e anaerobia. Il macrofago svolge questo lavoro metabolico per invaginare o estrudere nei vari piani dello spazio lembi citoplasmatici, onde procedere all'inglobamento di una o più particelle. In caso di particelle di piccole dimensioni (fino a 0,2 μm) l'ingestione avrebbe luogo quasi soltanto per un processo di invaginazione di membrana; in caso di particelle più voluminose (0,5-2 μm) essa conseguirebbe a una combinazione di invaginazione ed estroflessione di proiezioni citoplasmatiche. Quando debbano essere fagocitate particelle di grosse dimensioni (6-14 μm), il fenomeno avverrebbe esclusivamente per estroflessione di ampi lembi citoplasmatici: in quest'ultimo caso a volte il tentativo di ingestione completa fallisce, sebbene le particelle siano quasi completamente circondate dai lembi citoplasmatici del macrofago.
Si comprende, quindi, come il significato funzionale della fagocitosi sia quello di un meccanismo mediante il quale la cellula è in grado di aumentare le sue zone di contatto con una determinata superficie attraverso l'invaginazione della membrana o l'estroflessione di lembi citoplasmatici. Ciò premesso, si spiega anche un'importantissima caratteristica dei macrofagi: la capacità di aderire al vetro e ad altre superfici, fenomeno che può essere interpretato come il tentativo della cellula di fagocitare una sfera di diametro infinito (v. North, 1970; v. figg. 8 e 9).
Come abbiamo già ricordato, l'enorme energia richiesta dalla cellula fagocitaria per produrre il lavoro meccanico necessario a estendere i suoi lembi citoplasmatici, allo scopo di inglobare una o più particelle o di aderire al vetro, viene prodotta attraverso i processi glicolitici, che sembrano aumentare notevolmente in corso di fagocitosi. La successiva fase, cioè il completo inglobamento della particella in un vacuolo, non sembra invece richiedere grande consumo di energia da parte del macrofago.
A contatto con una cellula fagocitaria ogni singola particella è in grado di evocare un distinto fenomeno di fagocitosi, con formazione di un vacuolo contenente quindi una sola particella. Nella pinocitosi, invece, in un vacuolo sarebbero contenute più particelle, tutte molto piccole; d'altra parte, non solo le dimensioni di queste particelle sono troppo limitate per provocare ognuna la formazione di un vacuolo, ma sembra che le dimensioni del vacuolo stesso siano indipendenti da quelle della particella e quindi predeterminate dalle caratteristiche fisiologiche del macrofago.
Gli eventi successivi alla fagocitosi o alla pinocitosi consistono nel trasporto del materiale inglobato dalla periferia al centro del citoplasma. Durante questa fase il vacuolo fagocitario si fonde con i lisosomi primari o con lisosomi secondari preesistenti e ne riceve gli enzimi idrolitici necessari alla digestione della particella; dopo la digestione il vacuolo scompare lasciando al proprio posto un lisosoma secondario. Le vescicole pinocitiche si fondono fra di loro e ricevono le idrolasi dai lisosomi primari o dai corpi di Golgi, trasformandosi quindi anch'esse in lisosomi secondari (v. Cohn, 1970). L'azione concertata delle idrolasi acide lisosomiali porta alla trasformazione dei materiali biologici fagocitati in prodotti a basso peso molecolare, che possono quindi essere escreti o riutilizzati dal macrofago per fenomeni di biosintesi.
Evidentemente l'attività fagocitaria delle cellule del SRE costituisce il primo momento di una serie di eventi volti a eliminare dall'organismo materiali dannosi o inutili; in questo senso essa partecipa con un ruolo di primaria importanza a quei fenomeni di sorveglianza cellulare con cui l'organismo cerca di garantirsi una situazione di omeostasi.
Premesse queste note generali sulla fagocitosi macrofagica, descriveremo ora, sia pur succintamente, le principali funzioni del SRE.
b) Funzione antixenica.
Questa funzione del SRE, che noi preferiamo chiamare genericamente ‛di difesa', si esercita soprattutto attraverso la fagocitosi e consiste nell'eliminazione di materiali estranei penetrati nell'organismo (ad esempio particelle di carbone, di silice ecc.) e di numerosi microbi, virus e miceti. Ovviamente la massima attività fagocitaria viene espressa in condizioni normali dagli organi più ricchi di elementi macrofagici, quali il fegato e la milza; solo allorché la funzione di questi organi viene alterata da una loro grave compromissione, nel midollo osseo, nel polmone e nei tessuti connettivi ha luogo un'iperplasia compensatoria del SRE tesa a mantenere una normale attività fagocitaria nell'organismo. In altre condizioni tale attività può essere aumentata o depressa da un gran numero di fattori endogeni ed esogeni. Così, ad esempio, si può osservare un'attivazione del SRE in corso di infezioni batteriche, di malattie neoplastiche e di sindromi autoimmuni, quale espressione di una fisiologica risposta difensiva dell'ospite; al contrario, una depressione del SRE può essere presente in caso di insufficienza circolatoria, costituendo un fattore cruciale in caso di comparsa o di peggioramento di un'eventuale manifestazione patologica intercorrente.
La funzione difensiva del SRE è stata studiata sperimentalmente con estremo interesse: si è tentato, infatti, di stimolare il sistema per aumentare la resistenza alle infezioni e ai tumori, e si è tentato altresì di bloccarlo, o almeno inibirlo, per ritardare il rigetto di omotrapianti e per deprimere la risposta immune. Tra le principali sostanze stimolanti vi sono il glucano, lo zymosan, gli estrogeni, le endotossine e il BCG (Bacillus Calmette Guérin); tra quelle inibitrici il cortisone, i palmitati di metile e di etile e i sieri antilinfocitario e antimacrofagico. Tutte queste sostanze potrebbero esaltare, o inibire, la funzione del SRE, agendo tanto sulla fagocitosi pura e semplice, quanto sui meccanismi immuni a questa correlati. Infatti, oltre che attraverso la fagocitosi non immunitaria, il SRE svolge il suo ruolo antixenico anche attraverso processi fagocitari immunodipendenti.
Sebbene l'attività immunitaria propria dei macrofagi non sia ancora sufficientemente nota e numerosi siano i problemi insoluti, sappiamo oggi con certezza che queste cellule partecipano almeno a due momenti fondamentali della reazione immunitaria: al riconoscimento iniziale dell'antigene nel cosiddetto ‛braccio afferente' e alle manifestazioni dell'immunità cellulare nel cosiddetto ‛braccio efferente'.
L'interdipendenza fra macrofagi e linfociti sarebbe assai stretta: i primi, elementi aspecifici dell'immunità, non potrebbero operare in assenza dei secondi, elementi squisitamente specifici e quindi responsabili di una risposta mirata nei confronti dell'antigene volta a volta in causa. Nel ‛braccio afferente' della reazione immunitaria la presentazione dell'antigene da parte dei macrofagi ai T-linfociti stimolerebbe questi ultimi a produrre fattori capaci di attivare i macrofagi stessi, così da permettere loro di eliminare gli antigeni in causa. Contemporaneamente, i T-linfociti stimolati favorirebbero (helper function) la risposta anticorpale dei B-linfociti. Infine, l'interazione iniziale fra antigene, macrofagi, T-linfociti e B-linfociti trasformerebbe questi elementi linfatici in cellule dotate di memoria immune e quindi capaci, in caso di successivo contatto con l'antigene, di accelerare notevolmente la reazione immunologica (v. Katz, 1977; v. Unanue, 1980).
La partecipazione dei macrofagi ai meccanismi immuni non è limitata al riconoscimento iniziale dell'antigene e alla sua elaborazione, ma si estende al processo di immunità cellulare, cioè dell'ipersensibilità ritardata e dell'immunità protettiva. I modelli sperimentali dimostrano che i macrofagi giungono nelle sedi di reazione attratti da mediatori solubili, le ‛linfochine', prodotti da T-linfociti già sensibilizzati, ivi precedentemente pervenuti per effetto della stimolazione antigenica; successivamente i macrofagi sarebbero costretti a rimanere nel sito di reazione da un fattore di inibizione della loro migrazione (MIF, Migration Inhibitory Factor), prodotto anch'esso dai linfociti, e verrebbero quindi attivati, assumendo numerose caratteristiche che li distinguono dai macrofagi normali. Il macrofago attivato, di maggiori dimensioni e morfologicamente assai più complesso del macrofago classico, presenta un'esaltata attività fagocitaria, un maggior contenuto di idrolasi acide, una maggior capacità digestiva e un maggior indice mitotico. L'attivazione dei macrofagi risulterebbe in particolare indispensabile per limitare la virulenza e la sopravvivenza di quei batteri e parassiti che spesso vivono e si moltiplicano all'interno delle cellule fagocitarie, dove la loro esistenza non può essere messa in pericolo neanche da tassi elevatissimi di anticorpi serici battericidi. Già in passato Mackaness (v., 1964) aveva citato tutta una serie di microrganismi, il cui controllo potrebbe avvenire principalmente attraverso l'attivazione dei macrofagi: Bartonella, Leishmania, Trypanosoma, Plasmodium, Histoplasma, Mycobacterium, Brucella, Listeria, e altri ancora.
Oltre che nei confronti dei microrganismi citati, i macrofagi svolgerebbero un importante ruolo di difesa contro le infezioni virali: in cooperazione con i linfociti essi riuscirebbero infatti a distruggere, mediante meccanismi meramente citotossici, elementi cellulari infettati da virus. I macrofagi stessi potrebbero a loro volta essere infettati da virus (ad esempio virus vaccinico, virus herpetici), ma si tratterebbe di virosi abortive, utili a prevenire un'ulteriore diffusione dell'infezione.
Può essere considerata come facente parte delle funzioni di difesa la funzione di sorveglianza cellulare (‛omeocitostatica') e antineoplastica. Attualmente un grande interesse è dedicato allo studio della difesa dell'organismo contro doni abnormi caratteristici dei fenomeni di autoimmunità e a quello della molto discussa funzione antineoplastica. Indubbiamente l'organismo, per mantenere costante la sua struttura, deve disporre di meccanismi atti a neutralizzare le variazioni dell'informazione nucleica e del DNA, onde contrastare il pericolo di mutazioni somatiche accidentali. Il complesso di queste attività è variamente denominato: ‛meccanismi di mantenimento della costanza cellulare', ‛mechanisms for keeping mutations in check' (v. Koch e Miller, 1965), ‛omeocitostasi' e ‛meccanismi omeocitostatici' (v. Baserga, La regolazione..., 1961); la dizione più usata è quella burnetiana di ‛sorveglianza cellulare'. Per la polarizzazione dell'interesse degli studiosi sui componenti immunitari di tali meccanismi, si parla oggi spesso semplicemente di sorveglianza immunitaria, ma i meccanismi immunitari, anche se fra i più importanti, non sono i soli implicati; già noi ne avevamo elencati molti, fra i quali: minor fitness delle cellule trasformate, meccanismi biochimici enzimatici, strutture anatomiche localizzatrici, localizzazione delle mitosi in nicchie protette, ecc., oltre ai meccanismi immunitariosimili. Ed è fra questi meccanismi che si inserisce l'attività del sistema monocito-macrofagico, con l'intervento dei macrofagi documentabile, ad esempio, nel rigetto dei trapianti e nelle affezioni autoimmuni; in quest'ultimo caso, anzi, l'intervento dei macrofagi sembra ben più decisivo di quanto si riteneva in passato, dal momento che la funzione principale dell'ipersensibilità di tipo ritardato il cui ruolo è di così rilevante importanza in tali malattie - consisterebbe in questa ‛sorveglianza' (‛omeocitostasi'), che permette l'eliminazione di cellule o di ‛doni cellulari devianti' (ad esempio, cellule neoplastiche di qualsiasi origine antigenicamente alterate). La partecipazione dei macrofagi ai processi di immunità cellulare è dimostrata morfologicamente dal fatto che essi costituiscono il tipo cellulare predominante nelle reazioni di tipo ritardato (v. Nelson, 1969).
Un'altra azione svolta dai macrofagi nell'ambito dell'immunità cellulare sarebbe identificabile nella distruzione di cellule neoplastiche (v. Nelson, 1976), anch'essa mediata dalla loro attivazione, indotta aspecificamente o anche specificamente (v. Gill e Waller, 1977). Gli argomenti a favore di questo ruolo antineoplastico dei macrofagi derivano in massima parte da studi sperimentali condotti in vitro su cellule tumorali di animali. Numerosi fattori serici, o comunque ambientali, potrebbero interferire con questo ruolo distruttivo, aumentando o sopprimendo l'attività citotossica dei macrofagi (v. Hibbs e altri, 1977; v. Russell e McIntosh, 1977). A loro volta le cellule tumorali potrebbero mettere in atto meccanismi di autodifesa e abrogare la sorveglianza immunologica attraverso la produzione di inibitori della funzione macrofagica (v. Wood e Gollahon, 1977; v. Snyderman e altri, 1978). Sebbene l'ipotesi che i macrofagi uccidano cellule tumorali sia molto attraente, mancano ancora prove dirette di una soppressione tumorale nell'uomo mediata dai macrofagi: ciò nondimeno la massima parte dell'attuale interesse per l'immunoterapia nasce appunto dal tentativo di stimolare l'attivazione macrofagica nei confronti di cellule neoplastiche (v. Hopper e Pimm, 1976).
Il sistema reticoloendoteliale delle concezioni passate, e ugualmente il sistema monocito-macrofagico di quelle attuali, è sempre stato considerato soprattutto come un sistema particolarmente idoneo a svolgere funzioni di difesa dell'organismo, sia per la sua classica attività antixenica propriamente detta, sia per quella di sorveglianza cellulare omeocitostatica. In tal senso, una delle più rilevanti caratteristiche del sistema è rappresentata dalla sua adattabilità. Come noi sosteniamo da tempo, merita che si distinguano tessuti a rinnovamento selettivo e altri a rinnovamento aselettivo; il SRE, dotato di un'alta potenzialità riproduttiva che conserva anche dopo la migrazione dei monociti nei siti dove sono chiamati a esplicare la loro attività, rappresenta un tipico esempio di sistema caratterizzato da riproduzione spiccatamente selettiva, con possibilità di prevalente proliferazione dei doni cellulari interessati in senso burnetiano e con alta adattabilità. Nel 1961 avevamo scritto: ‟Il sistema delle cellule istiocitarie migranti deve esplicare una serie di funzioni difensive varie da situazione a situazione, deve cioè essere ‛adattabile'. Ci pare meritevole di esser tenuta presente l'ipotesi che uno dei meccanismi più preziosi di questa adattabilità delle cellule RI sia proprio questa loro crescita selettiva con l'imporsi selettivo dei vari doni a diversa impronta genica a seconda degli occasionali bisogni dell'organismo" (v. Baserga, La regolazione..., 1961).
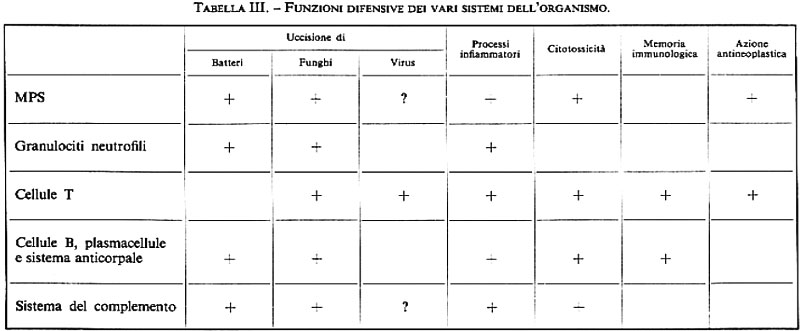
La funzione di difesa dell'organismo è la principale tra quelle attribuite al SRE, ma a essa, come risulta sempre più evidente dal progresso delle conoscenze, partecipano certamente anche altri sistemi. Nella tab. III, che ne ricalca una recente di Kleihauer (v., 1978), sono esposte sinteticamente le concezioni attuali.
c) Funzione citocateretica.
Questa funzione, strettamente dipendente dall'attività fagocitaria, costituisce un momento di grande importanza nel mantenimento dell'omeostasi organismica. Le cellule del SRE possiedono infatti la capacità di fagocitare e di distruggere cellule senescenti o morte. Loutit (v., 1960) ricordava infatti che, mentre le cellule degli epiteli di rivestimento cutaneo-mucosi vengono eliminate per sfaldamento, le cellule degli organi interni, quando sono giunte al termine del loro ciclo vitale per senescenza o per cause patologiche, vengono fagocitate da elementi appartenenti al SRE.
La funzione citocateretica del sistema si manifesta con massima evidenza nella rimozione e distruzione delle cellule del sangue (ematoclasia): globuli rossi, globuli bianchi e piastrine, invecchiati o alterati, in condizioni normali o patologiche, vengono pressoché tutti fagocitati dal SRE splenico, epatico, midollare e/o generalizzato. La principale funzione in questo senso sembra essere quella svolta dal SRE splenico: è prevalentemente nella milza, ad esempio, che vengono intrappolati e distrutti gli sferociti, gli eritrociti danneggiati da anticorpi, forse anche quelli giunti al termine del loro ciclo vitale, in definitiva i globuli rossi che non rispondono a un minimo di requisiti essenziali (la cosiddetta culling function: v. Crosby, 19772). Il reticoloendotelio splenico permette inoltre la rimozione di tutta una serie di inclusioni eritrocitarie patologiche (corpi di Jolly, corpi di Heinz, granuli di ferro, granuli di Minnich e simili) che possono compromettere la normale funzione e sopravvivenza eritrocitaria (la cosiddetta pitting function di Crosby). Analogamente il SRE, e soprattutto quello splenico, può rimuovere un gran numero di leucociti neutrofili danneggiati dalle endotossine, gli elementi contenenti inclusioni superficiali come i corpi di Foà-Kurloff, e i linfociti danneggiati da varie noxae linfolitiche (raggi X, citostatici, cortisonici, ecc.; v. fig. 10).
In caso di estrema necessità (imponente emolisi extravascolare, ecc.) le funzioni citocateretiche possono essere assunte prevalentemente dal fegato, e in caso di splenectomia il SRE extrasplenico può compensare, almeno in parte, le funzioni della milza.
d) Funzione emopoietica.
A tutt'oggi, nonostante la gran mole di ricerche condotte sull'argomento, non si è giunti ancora alla definitiva identificazione delle cellule staminali capaci di dare origine ai vari elementi del sangue. Molti autori hanno ritenuto di poter attribuire tale funzione emopoietica a elementi reticoloendoteliali (v. Barnes e Loutit, 1967), in accordo con la concezione classica della scuola ematologica italiana, da A. Ferrata in poi, che vedeva nell'emoistioblasto la cellula capostipite di ogni elemento figurato del midollo osseo e del sangue periferico.
A prescindere dal grande problema dell'identificazione della cellula staminale, è certo comunque che i macrofagi partecipano all'emopoiesi, producendo almeno quattro fattori capaci di stimolarla o di modularla mediante meccanismi di feedback positivi o negativi: 1) il CSF (Colony Stimulating Factor), capace di stimolare la cellula progenitrice mieloide a produrre colonie granulomonocitarie; 2) l'eritropoietina, capace di indurre specificamente la differenziazione di precursori eritroidi in elementi eritroidi riconoscibili; 3) il BPA (Burst Promoting Activity), in grado di promuovere il rapido sviluppo di colonie dopo un periodo iniziale di riposo o di lenta proliferazione; 4) la PGE2 (prostaglandina E2), che sembra indurre la CFU-S (Colony Forming Unit-Spleen, unità capace di formare colonie spleniche) a entrare in fase proliferativa, ma può anche limitare un'eccessiva proliferazione mieloide (v. Kurlande altri, 1978; v. Cime e Golde, 1979; v. Peschle, 1980). il fatto che nella produzione di alcuni di questi fattori emopoietici (BPA e CSF) sia indispensabile la compartecipazione dei T-linfociti sarebbe un'ulteriore prova degli strettissimi rapporti esistenti fra MPS e sistema linfoide.
e) Funzione trofica.
Questa funzione del SRE, più postulata che dimostrata, si basa su due reperti istologici e citologici caratteristici: l'isolotto reticoloeritroblastico e l'isolotto reticololinfocitario. Le cellule reticoloendoteliali sistemate al centro di ammassi di elementi eritroblastici e linfocitari apporterebbero a questi ultimi materiali nutritizi e/o molecole dotate di informazioni necessarie alle loro funzioni.
L'isolotto reticoloeritroblastico, reperibile a livello del midollo osseo, è composto di una o due cellule ‛reticolari' (di fatto macrofagiche) circondate da una corona di eritroblasti in vari stadi di maturazione e di reticolociti (v. fig. 11). Come osserva Bessis (v., 1972), questi macrofagi, cosiddette ‛cellule nutrici', giocherebbero un ruolo attivo in momenti diversi della vita degli eritroblasti e dei globuli rossi, fagocitando i nuclei eritroblastici, una volta estrusi, e i globuli rossi giunti al termine della loro esistenza, ma anche fornendo agli eritroblasti sostanze necessarie alla loro nutrizione, maturazione e omeostasi, e forse anche eritropoietina e ferro. In condizioni normali gli isolotti reticoloeritroblastici sono rari, ma in certe anemie emolitiche acute e soprattutto nelle eritremie croniche il loro numero aumenta sensibilmente, tanto da rivestire un certo significato diagnostico (v. Di Guglielmo, 1943 e 1946).
Nel caso dell'isolotto reticololinfocitario, la cellula macrofagica sarebbe circondata da linfociti (cosiddetta ‛peripolesi'). Che fra i due tipi cellulari vi siano rapporti stretti è dimostrato dalla possibilità di temporanee adesioni citoplasmatiche fra linfociti e macrofagi a mezzo di recettori macrofagici non specializzati (v. Rosenthal e altri, 1975), ma non si sa ancora con certezza quali possano essere le sostanze trasferite dal macrofago al linfocito nell'ambito dell'isolotto. Potrebbe trattarsi di materiali di immediata utilità trofica per i linfociti (v. Chen e Hirsch, 1972; v. Mosier e Pierce, 1972), così come di antigeni o di acidi nucleici antigenicamente informati. Secondo Rosenthal e altri (v., 1975) l'interazione fisica fra macrofago e linfociti sarebbe un fenomeno inizialmente indipendente dall'antigene; qualora però il macrofago fosse portatore di un antigene per il quale il linfocito già possiede un recettore immunospecifico, ne seguirebbe l'attivazione della DNA-sintesi linfocitaria, a documentazione di una risposta immunitaria specifica.
f) Funzione metabolica.
Un resoconto dettagliato di tutte le attività metaboliche, cui il SRE partecipa in qualche modo, sarebbe assai difficile, dal momento che i macrofagi intervengono in un numero enorme di eventi biochimici e fisiologici. Inoltre si deve tener presente che solo negli ultimi anni è ripreso lo studio approfondito del SRE, e che pertanto è molto probabile che in futuro si potrà dimostrare la sua partecipazione in attività fisiologiche finora non sospettate. Pur con queste limitazioni, un esame della letteratura mostra come il SRE partecipi alle seguenti funzioni metaboliche di estrema importanza (v. Saba, 1970; v. Cline, Biochemistry and..., 19772; v. Meuret, 1977; v. Nathan e altri, 1980): a) produzione di fattori chemiotattici come componenti del complemento, attivatore del plasminogeno, enzimi lisosomiali, collagenasi ed elastasi, prostaglandine e altri ancora (dalla produzione di alcuni di questi fattori dipendono certe funzioni descritte qui di seguito); b) rimozione dal sangue, per mezzo della fagocitosi, di materiali tessutali autologhi al termine del loro ciclo vitale; c) rimozione di proteine denaturate e metabolismo delle proteine; d) rimozione e detossificazione delle endotossine e di altri composti tossici; e) rimozione di microaggregati di fibrina, e quindi prevenzione della coagulazione intravascolare; f) rimodellamento delle ossa; g) metabolismo del ferro e formazione di bilirubina per mezzo della fagocitosi di eritrociti; h) metabolismo degli steroidi; i) trasformazione biologica ed escrezione del colesterolo; 1) metabolismo e detossificazione dei farmaci; m) produzione di interferon.
5. La patologia del sistema reticoloendoteliale.
a) Premesse.
L'inquadramento nosologico delle malattie del SRE risente dei rimarchevoli progressi occorsi in questi ultimi anni nell'area della fisiopatologia del SRE e del sistema linfatico. Le nuove conoscenze su distribuzione, origine, cinetica e ruolo degli istiociti e dei linfociti hanno completamente rinnovato la nosologia delle malattie del SRE. Alcune malattie che in passato venivano considerate tipiche affezioni del SRE, ad esempio la micosi fungoide, la sindrome di Sézary e perfino il reticulum cell sarcoma, sono state riesaminate e attribuite, sulla base di concezioni più moderne e di ragionevoli prove clinico-sperimentali, a disordini del sistema linfatico (v. sangue: Leucemie). D'altra parte le migliori conoscenze che abbiamo oggi sull'istiocito ci permettono di classificarne la patologia su basi che non sono più meramente morfologiche, come in passato, ma che tengono conto delle caratteristiche funzionali della serie istiocitaria.
b) Le classificazioni tradizionali.
L'interpretazione del SRE come unità funzionale, sostenuta da Aschoff, ha influenzato a lungo patologi e clinici, dando luogo - come già si è detto - a tentativi di costruzione di una teoria unitaria delle malattie del SRE e a inevitabili eccessive generalizzazioni: entrarono quindi a far parte del nuovo edificio nosografico una quantità enorme di quadri morbosi, accomunati solo dall'indubbia compartecipazione del SRE, anche se in funzione secondaria.
Riteniamo opportuno nella presente trattazione parlare solo delle forme patologiche primitive del SRE, senza soffermarci sulle iperplasie che si riscontrano in corso di infezioni, infestazioni, iperemolisi, cioè sulle cosiddette reticoloendoteliosi secondarie, il cui studio peraltro è stato molto utile in passato per acquisire migliori conoscenze sulla morfologia e sulla distribuzione del SRE. Così pure non ci soffermeremo sulle reticoloendoteliosi da tesaurismosi, come le sfingolipidosi e le mucopolisaccaridosi, caratterizzate da deficit enzimatici, che impediscono il normale catabolismo dei lipidi e dei mucopolisaccaridi, e/o da un'iperproduzione cellulare eccedente le fisiologiche capacità di fagocitosi possedute da istiociti normali. Ci basti comunque, a questo proposito, riportare la classificazione delle malattie del SRE proposta da Di Guglielmo e Baserga (v., 1952) e successivamente ripresa da Baserga (v., Generalità..., 1961). Essa fa il punto sulla posizione nosografica di queste malattie poco prima dell'introduzione del concetto di MPS.
1. Reticoloendoteliosi da alterata funzione del ricambio del sangue: a) reticoloendoteliosi da iperattività citoemopoietica (monocitosi secondarie e monocitosi primitive o leucemie monocitiche; endoteliosi secondarie come l'endocardite lenta, la malaria, il kala-azar, ecc.; endoteliosi primitive ed emoistioblastosi con apparizione in circolo di emoistioblasti); b) reticoloendoteliosi da ipofunzione emopoietica (è un gruppo ipotetico, cui non si possono ancora ascrivere casi sicuramente dimostrati); c) reticoloendoteliosi da iperattività emocateretica (ittero emolitico); d) reticoloendoteliosi da ipoattività emocateretica.
2. Reticoloendoteliosi da alterata funzione del ricambio dei grassi e dei lipoidi: a) secondarie (iperplasia lipoidocellulare diabetica di Siegmund e Fahr, granulomatosi xantomatosa di Kirsch, ecc.); b) primitive (malattie di Gaucher, di Niemann-Pick, di Hand-Schüller-Christian).
3. Reticoloendoteliosi sistemiche: a) secondarie o sintomatiche (sepsi, malaria, leishmaniosi, ecc.); b) primitive (come nei casi di Goldschmidt e Isaac, ecc.); c) neoplasie del SRE (reticoloendoteliomi).
4. Reticoloendoteliosi infiammatorio-proliferative (granulomi): a) granulomi a eziologia nota (tubercolari, actinomicotici, ecc.); b) granulomi a eziologia sconosciuta (linfogranuloma maligno di Sternberg).
I problemi più rilevanti circa l'inquadramento delle malattie del SRE sono sorti però da quando questo cominciò a essere studiato non solo con i classici criteri morfologici, basati sulla comune osservazione microscopica, ma anche con metodi funzionali. Precedentemente, infatti, l'attribuzione alla patologia del SRE di un determinato quadro morboso era affidata al riconoscimento di aspetti istologici e citologici, ben codificati dall'ematologia classica, ma non ancora sostenuti da una conoscenza sufficiente delle funzioni e delle proprietà dei linfociti e degli istiociti. Negli ultimi anni sono state proposte numerose tecniche funzionali atte a permettere la distinzione fra i vari sottogruppi linfocitari ed elementi appartenenti al SRE (monociti e macrofagi). Lo sviluppo di questi studi funzionali sembra oggi escludere l'appartenenza alla patologia reticoloendoteliale di malattie tradizionalmente ritenute proliferazioni maligne del SRE, come la malattia di Sézary e la micosi fungoide, i cui tessuti patologici sembrerebbero in realtà essere costituiti da T-linfociti. Analogamente il classico reticolosarcoma ‛scarsamente differenziato' viene quasi unanimemente ritenuto di natura linfocitaria (v. Lukes e Collins, 1974): le grandi dimensioni, la presenza del nucleolo e l'iperbasofilia citoplasmatica, caratteristiche delle cellule di questa forma ‛reticolosarcomatosa', sono comuni anche ai tipici immunoblasti da PHA (fitoemoagglutinina) e da PM (Pokeweed Mitogen); la presenza o meno di elementi plasmacellulari d'accompagnamento nei singoli casi costituirebbe poi un reperto a favore della natura linfocitaria (B, T o null) di questi tumori immunoblastici.
c) Le nuove classificazioni proposte.
Forse non è ancora possibile procedere a una revisione della sistemazione nosologica delle malattie del SRE e quindi a una loro definitiva classificazione, dal momento che la letteratura ematologica presenta di giorno in giorno un affastellarsi di dati in proposito, spesso incerti o addirittura contraddittori. Nondimeno alcuni autori hanno proposto nuove classificazioni delle malattie del SRE che, seppur non esenti da critiche, possono attualmente costituire lo strumento per ulteriori fattive indagini e discussioni. Ci riferiamo in particolare alle classificazioni di Meuret (v., 1977) e di Cline e Golde (v., 1973). Nella sua classificazione, riportata nella tab. IV, Meuret allarga smisuratamente i confini delle malattie del SRE, che suddivide in quattro gruppi costituiti, come egli stesso afferma, da ‟un pot-pourri di entità morbose diverse". Per i motivi riportati nelle pagine precedenti, non ci sentiamo di condividere tale tipo di classificazione: la compartecipazione del SRE alla patologia di altri tipi cellulari o di precursori comuni deve sempre essere tenuta presente, ma non deve far trascurare il ruolo preminente che l'alterazione di altri sistemi riveste nella storia naturale delle singole malattie.
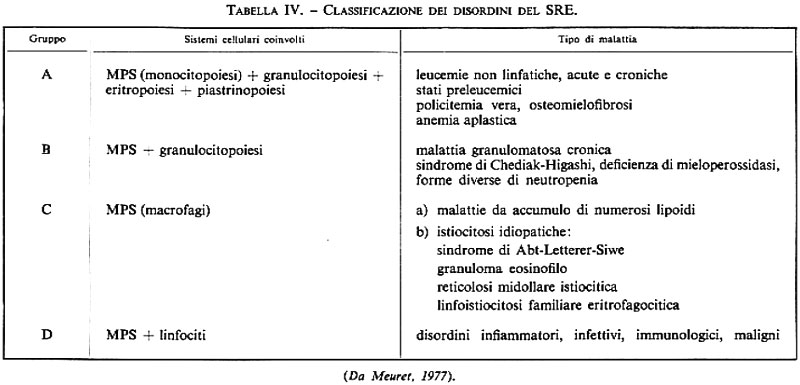

Noi pensiamo che la classificazione dei disordini istiocitari proposta da Cline e Golde si attagli più di quella di Meuret alla realtà anatomoclinica delle malattie del sistema monocito-macrofagico, e quindi a essa ci atterremo nella nostra trattazione. Nella tab. V riportiamo senz'altro la classificazione dei disordini del SRE secondo Cline e Golde, sia pure con una lieve modificazione (l'aggiunta della reticolosi eritrofagocitica familiare) e con un'appendice concernente l'hairy cell leukemia. Appare evidente che questa classificazione considera le malattie del SRE come una serie di quadri clinici sostenuti da disordini proliferativi di cellule in vario grado di differenziazione, da quelle altamente indifferenziate della leucemia monocitica a quelle mature dell'istiocitoma localizzato. Tratteremo succintamente queste malattie.
d) Malattie del SRE con differenziazione cellulare scarsa o variabile.
1. Leucemia monocitica. - Fin dal 1913, dopo la prima, e peraltro discussa, osservazione di Reschad e SchillingTorgau, cui seguirono l'anno successivo i casi di Fleischmann e di Castellino e Ferrata, erano note forme leucemiche con presenza in circolo di cellule più o meno immature e atipiche, riconducibili alla serie monocitaria.
Inizialmente Reschad e Schilling-Torgau (v., 1913) avevano parlato di ‛leucemia a splenociti' (Spienozyten-Leukamie), indi Schilling aveva coniato il termine di ‛reticoloendoteliosi leucemica', ma poi era prevalsa la denominazione di ‛leucemia monocitica'. Il problema dell'interpretazione di queste forme aveva comunque dato luogo, anche in un passato lontano (v. Baserga, 1933, per la vecchia letteratura), a vive discussioni. Quando si riteneva che i monociti avessero un'origine endoteliale, indipendente dalle cellule mieloidi, alcuni interpretavano la leucemia monocitica come l'espressione di una forma a sé, reticoloendoteliale; altri, invece, come Naegeli, che aveva sempre ricondotto il monocito alla serie mieloide, o negavano addirittura l'esistenza delle leucemie monocitiche, o identificavano queste forme come una varietà di leucemia mieloblastica (paramieloblastica). Poiché la compartecipazione mieloide in molte leucemie monocitiche era evidente, si erano classificate forme tipo Schilling, o reticoloendoteliali propriamente dette, e forme tipo Naegeli, o mieloidi; si ammetteva anche l'esistenza di forme miste. Le forme croniche, molto più rare delle acute, venivano in genere considerate come reticoloendoteliali in senso stretto.
A seguito della dimostrazione, accettata da quasi tutti gli autori, che il monocito è una cellula di derivazione midollare, le leucemie cosiddette monocitiche sono ormai considerate come forme mieloidi. Inoltre la ripetuta constatazione che gli elementi d'aspetto monocitico o monocitoide presenti in queste leucemie acute sono raramente isolati, ma quasi sempre accompagnati da elementi mieloblasticopromielocitari, consente di affermare che gran parte dei casi devono essere considerati come forme mielomonocitiche. Occorre ricordare poi che una componente di cellule d'aspetto ‛endoteliale' costituisce un reperto frequente anche nei casi di mielosi eritremica, fenomeno già descritto da Di Guglielmo, il cui significato è attualmente chiaro alla luce dell'accertata comune origine mieloide di entrambe le serie.
Naturalmente le più recenti indagini hanno permesso un'ulteriore identificazione dei vari tipi morfologici della leucemia monocitica, superando i criteri quasi esclusivamente morfologici utilizzati in passato.
Le indagini citochimiche, più specifiche, lo studio dell'attività muramidasica serica e cellulare e il test del nitro-blu-tetrazolio (NBT) hanno permesso a Catovsky e Galton (v., 1973) di proporre un'ulteriore classificazione delle leucemie monocitiche: a) leucemia mielomonocitica acuta di tipo II, caratterizzata da un'elevata concentrazione di lisozima serico e dal reperto di un gran numero di cellule blastiche capaci di produrre lisozima (in media il 77%), di ridurre il NBT (in media il 33%) e di mostrare un'intensa attività esterasica non specifica; b) leucemia monoblastico-monocitica o monocitica acuta (v. fig. 12), anch'essa caratterizzata da numerosi blasti capaci di produrre lisozima (in media il 61%), di ridurre il NBT (in media il 31%) e di dare positività alla reazione per le esterasi non specifiche, e inoltre da un tasso assai elevato di lisozima serico; c) leucemia monocitica cronica, caratterizzata da un'elevatissima percentuale di cellule capaci di produrre lisozima (fra il 90 e il 100%), il cui numero è correlabile con il contenuto serico di questo enzima e con il tasso di elementi NBT-positivi (che assommano in media al 33%); tali cellule risultano negative alla reazione per le mieloperossidasi.
La leucemia mielomonocitica acuta di tipo I, invece, andrebbe considerata, a tutti gli effetti, come una vera leucemia mieloide acuta, dal momento che in questa forma è reperibile un numero inferiore di elementi blastici lisozima e NBT-positivi (rispettivamente il 22,5% e il 6,7%) e un ridotto tasso serico di lisozima. Va ricordata inoltre, quale test particolarmente utile per distinguere le forme monocitiche da quelle mieloidi, la metodica, applicata sullo stesso vetrino, della doppia incubazione per la dimostrazione della naftol-ASD-acetato-esterasi fluoruro sensibile e della naftol-ASD-cloroacetato-esterasi, le cui positività indicherebbero rispettivamente la natura monocitica o mieloide delle cellule in studio. Questi test diagnostici permettono ormai la netta e definitiva distinzione fra il gruppo delle leucemie monocitiche e quello delle leucemie mieloidi acute tipiche.
Dal punto di vista anatomopatologico, le leucemie monocitiche non si distinguono macroscopicamente dalle altre leucemie acute e generalmente vengono descritte insieme a queste (v. Grusovin, 1974). Gli organi costantemente coinvolti sono ovviamente, oltre al midollo osseo, la milza, i linfonodi e il fegato; viene inoltre segnalata, come caratteristica della leucemia monocitica, la comparsa di una gengivopatia produttiva. Istologicamente si osservano il sovvertimento strutturale dei linfonodi e della milza, la presenza di numerose mitosi e, occasionalmente, fenomeni di eritrofagocitosi.
Il quadro clinico, analogamente a quello anatomopatologico, non permette in genere di distinguere la leucemia monocitica dalle altre leucemie acute. Si osserva generalmente, accanto ad altri sintomi generici, la prevalenza di una sindrome emorragica da piastrinopenia, di infezioni recidivanti con granulocitopenia, di uno stato anemico e di lesioni d'organo o d'apparato. Fra queste ultime orienteranno in particolare verso una leucemia monocitica la tumefazione delle gengive e una sofferenza dei denti (osservabili, peraltro, anche nella leucemia mieloblastica acuta), lesioni cutanee, papulose o nodulari, isolate o confluenti, un'eventuale nefropatia tubulare da lisozimuria e il precoce, ma non frequente, interessamento porotico delle ossa.
La malattia presenta generalmente un decorso più o meno acuto e ha esito letale entro un anno dalla diagnosi, anche se sono stati segnalati pazienti sopravvissuti per tempi compresi fra i quattro e gli undici anni. Wintrobe (v., 1973) considera tali lunghe sopravvivenze come caratteristiche della variante cronica della leucemia monocitica.
Le possibilità terapeutiche nei confronti di queste forme appaiono piuttosto limitate: i farmaci più efficaci parrebbero un derivato dell'epipodofillotossina (VP 16.213) e una antraciclina (rubidazone), utilizzati da soli, in abbinamento, o in associazione con citostatici di meno recente impiego, come la daunomicina, l'arabinoside C e la σ-tioguanina.
2. Reticulum cell sarcoma o linfoma istiocitico. - Questo disordine del SRE viene tradizionalmente inquadrato fra i linfomi non Hodgkin, come si può constatare nelle classificazioni istologiche di Gall e Mallory (v., 1942), di Rappaport e altri (v., 1956) e di Rappaport (v., 1966). Secondo l'anatomia patologica classica, esso costituirebbe il 20% di tutti i linfomi non Hodgkin, ma un recente approccio funzionale alla classificazione dei linfomi maligni, operato da Lukes e Collins (v., 1974), tenderebbe a escludere la natura reticoloendoteliale della gran parte dei linfomi istiocitici secondo Rappaport. Infatti è stato dimostrato che la massima parte dei cosiddetti reticolosarcomi, e particolarmente quelli scarsamente differenziati, sono di origine linfocitaria. Lukes e Collins pongono alla base del loro inquadramento dei linfomi maligni la distinzione degli elementi linfatici in linfociti di tipo B (bursaequivalenti, timoindipendenti), T (timodipendenti) e U (linfociti indefiniti), e attribuiscono la gran parte dei linfomi a una compromissione patologica di questi tre tipi cellulari. Naturalmente i linfomi di tipo istiocitico non possono essere esclusi del tutto dai più moderni inquadramenti ma, come abbiamo già detto, la massima parte dei reticolosarcomi classici vengono ormai interpretati come linfomi di cellule B, T, o U, più spesso delle prime. La frequenza dei linfomi d'origine monocitomacrofagica risulterebbe pertanto compresa fra l'1 e il 5% della totalità dei linfomi non Hodgkin (v. Lukes e altri, 1978; v. Nathwani e altri, 1978), e non intorno al 20% come in precedenza si riteneva.
In conseguenza dell'ancora incerta sistemazione nosografica, il quadro anatomopatologico del reticolosarcoma non è chiaramente delineato. I reperti non sono distinguibili dagli altri linfomi non Hodgkin: possono infatti osservarsi localizzazioni ai linfonodi superficiali e profondi, con maggiore frequenza a quelli cervicali e mediastinici, e rapide diffusioni successive ad altre stazioni (v. fig. 13). Nelle forme diffuse, la generalizzazione avviene di regola a tappe successive (v. Lanza, 1974). I linfonodi colpiti si presentano più o meno aumentati di volume e tendono ad aderire fra di loro e con i tessuti vicini. Spesso la sede primitiva del reticolosarcoma sembra essere extralinfonodale; le localizzazioni extralinfonodali sono comunque assai comuni, soprattutto frequenti a livello delle tonsille e del rinofaringe, della milza, del tratto gastroenterico, delle ossa, più raramente nella tiroide, nei testicoli, nell'apparato genitourinario e nel polmone (v. Cline e Golde, 1973).
Sul riconoscimento istologico dei reticolosarcomi regna ora la massima incertezza. Precedentemente se ne distinguevano due varietà principali, una forma sinciziale indifferenziata e una varietà istiocitica differenziata (v. Wintrobe, 1973): in quest'ultima era comune il riscontro di fenomeni di fagocitosi nei confronti di globuli rossi, cellule nucleate e débris cellulare. Ulteriori delucidazioni potranno conseguire al progresso delle conoscenze sui rapporti fra morfologia classica, funzioni e markers immunologici.
Dal punto di vista clinico la differenziazione fra reticolosarcoma e altri linfomi non Hodgkin appare di estrema difficoltà, anche se una localizzazione primitiva nei linfonodi addominali o in sede extralinfonodale è assai più frequente nel primo che nei secondi. Analogamente, nel reticolosarcoma sono assai più frequenti le lesioni di tipo osteolitico, soprattutto in caso di malattia generalizzata. Per il resto il quadro clinico è sovrapponibile a quello degli altri linfomi e si distingue da quello del morbo di Hodkgin per la maggior frequenza di sintomi generali ed eventualmente di risentimenti pleurici, per un più comune interessamento dell'anello di Waldeyer, del tubo gastroenterico e dei linfonodi mediastinici e un meno frequente interessamento del midollo osseo, dei linfonodi mesenterici, della milza e probabilmente del fegato. Il reticolosarcoma inizia spesso all'interno dell'addome, presenta frequentemente un'invasività locale e quindi comporta più comunemente una localizzazione extralinfatica per estensione.
Il decorso della malattia tende a essere aggressivo, consentendo una sopravvivenza media di circa un anno; può rispondere alla radioterapia e alla chemioterapia e, in caso di remissione, può consentire una sopravvivenza significativamente lunga (v. Skarin e Canellos, 1980).
e) Malattie del SRE con differenziazione cellulare moderata.
1. Malattia di Abt-Letterer-Siwe (istiocitosi acuta differenziata secondo Rappaport). - Questa affezione viene da tempo considerata come la forma acuta diffusa della cosiddetta ‛istiocitosi X' (v. Lichtenstein, 1953), grande entità nosologica comprendente anche forme diffuse croniche (malattia di Hand-Schüller-Christian) e forme localizzate (granuloma eosinofilo dell'osso o extraosseo, e istiocitosi focale). La malattia di Abt-Letterer-Siwe è caratterizzata dalla proliferazione di istiociti moderatamente differenziati in vari organi, ma particolarmente nella cute, nei linfonodi, nella milza e nel midollo osseo. L'aspetto istologico delle lesioni può ricordare quello del reticolosarcoma ben differenziato; le cellule patologiche presentano in genere un nucleo ovalare o reniforme e abbondante citoplasma acidofilo non contenente lipidi; a volte possono essere presenti in proporzioni variabili istiociti contenenti lipidi o altri materiali fagocitati. I rapporti con altre affezioni istiocitarie sembrerebbero documentati dal trapasso di questa reticolosi in altre forme di istiocitosi (v. Lanza, 1974) e perfino in leucemie monocitiche.
La malattia di Abt-Letterer-Siwe, che si manifesta preferibilmente nei bambini al di sotto dei due anni, si presenta dal punto di vista clinico come un'affezione grave, febbrile, caratterizzata da: 1) alterazioni cutanee papulose, maculopapulose e purpuriche, che prediligono il torace e il viso; 2) costante epatosplenomegalia associata eventualmente a ittero, ascite e/o segni di ipertensione portale; 3) linfoadenomegalia a carico delle stazioni superficiali, per compartecipazione dei linfonodi all'affezione sistemica, ma anche come espressione di una loro reattività alla superinfezione delle lesioni cutaneee; 4) compromissione del parenchima polmonare (dal 10 al 50% dei casi), con aspetti miliari, microreticolonodulari, nodulari o chiaramente broncopneumonici (v. Schaison e Seligman, 1972); 5) lesioni ossee lacunari costantemente prive di reazione periostale a carico del cranio (con particolare frequenza a livello della volta, della mastoide o delle pareti orbitarie), di varie ossa piatte (scapole, coste superiori e bacino) e, più di rado, delle ossa prossimali degli arti.
L'evoluzione della malattia si compie a poussées successive caratterizzate da elevazione termica, estensione delle lesioni cutanee e aggravamento della sintomatologia respiratoria; le più comuni complicazioni fatali sono costituite dalle emorragie e dalle infezioni. Tuttavia la malattia non è sempre letale: il 30% dei pazienti può presentare un'evoluzione verso una forma meno diffusa, o una remissione di entità variabile, o addirittura la guarigione (v. Doede e Rappaport, 1967).
2. Reticolosi midollare istiocitica (istiocitosi maligna secondo Rappaport). - La posizione nosografica di questa affezione è stata oggetto di numerose discussioni. Ricordiamo come il termine sia stato proposto da Scott e RobbSmith (1939) per indicare la proliferazione focale di istiociti nella porzione midollare dei linfonodi: la mancanza di manifestazioni ematiche e i rilevanti fenomeni di eritrofagocitosi caratterizzerebbero ulteriormente la reticolosi midollare istiocitica, sebbene anche in questa forma si possa occasionalmente osservare la comparsa nel sangue di cellule abnormi e, d'altra parte, l'eritrofagocitosi sia rilevabile anche nel reticolosarcoma e nella leucemia monocitica. Dal punto di vista citologico, gli elementi caratteristici di questo disordine sembrerebbero istiociti abbastanza differenziati con elevata capacità proliferativa e fagocitaria, per cui alcuni hanno ritenuto di poter identificare questa malattia con la forma ben differenziata del reticolosarcoma (v. Cline e Golde, 1973).
Queste osservazioni, che parrebbero dimostrare la natura reticoloendoteliale delle cellule patologiche caratteristiche della reticolosi midollare istiocitica, hanno ricevuto conferma negli anni a noi piu vicini da indagini ultrastrutturali, citochimiche, immunologiche e funzionali (v. Huhn e Meister, 1978; v. Jaffè e altri, 1975; v. Mendelsohn e altri, 1980), sicché la posizione nosografica di questa malattia appare finalmente chiarita e la sua inclusione nella patologia del SRE definitivamente accertata.
L'aspetto istologico caratteristico della reticolosi midollare istiocitica è costituito dalla distribuzione delle cellule neoplastiche nei seni midollari dei linfonodi, nella polpa rossa splenica, nei sinusoidi epatici e in focolai sparsi irregolarmente all'interno del midollo osseo. Le alterazioni iniziali appaiono confinate ai sinusoidi, così che l'architettura degli organi colpiti è spesso abbastanza ben conservata, nonostante la proliferazione massiva di cellule neoplastiche. Spesso l'aspetto degli istiociti patologici appare ingannevolmente benigno, così da simulare un'istiocitosi reattiva dei seni, ma altre volte esso risulta chiaramente abnorme, e in una minoranza di casi può essere pleiomorfico e bizzarro, rendendo allora difficile la diagnosi differenziale con il morbo di Hodgkin, con il reticolosarcoma pleiomorfico, o addirittura con il carcinoma anaplastico. Gli istiociti meglio differenziati mostrano un'attività fagocitaria superiore a quella dei meno differenziati, attività che si svolge soprattutto nei confronti degli eritrociti, ma anche di detriti cellulari, lipidi, emosiderina o altri materiali. La fagocitosi di elementi del sangue da parte degli istiociti può contribuire all'instaurarsi dell'anemia e della pancitopenia caratteristiche della reticolosi midollare istiocitica.
Si tratta di una malattia rara che colpisce soggetti adulti di entrambi i sessi e che presenta decorso rapidamente progressivo e fatale, con una sopravvivenza variabile da 1 a 15 mesi, mediamente inferiore ai 6 mesi. Clinicamente essa è caratterizzata da febbre, deperimento, linfoadenomegalia, epatosplenomegalia, pancitopenia progressiva, talora anche da ittero e da porpora. Talvolta può essere presente un'enteropatia protidodisperdente, provocata dall'infiltrazione istiocitaria della parete intestinale. In alcuni casi, contraddistinti da notevole splenomegalia, il quadro clinico e anche quello istologico possono simulare la reticoloendoteliosi leucemica. Il trattamento della malattia con cortisonici e citostatici appare del tutto insoddisfacente, potendone sortire soltanto risposte di assai breve durata.
3. Reticolosi eritrofagocitica familiare (o linfoistiocitosi eritrofagocitica familiare). - Si tratta di una malattia familiare dei sistemi linfoide e reticoloendoteliale, che colpisce bambini d'ambo i sessi, spesso in età nipiologica (v. Farquhar e Claireaux, 1952); la malattia sembra trasmessa come carattere autosomico recessivo, sebbene in alcuni casi non si siano potute documentare le modalità di trasmissione.
Il quadro istopatologico ricorda quello della reticolosi midollare istiocitica nella sua forma meno atipica, con prevalenza dei fenomeni fagocitici rispetto a quelli proliferativi. La somiglianza fra la reticolosi eritrofagocitica familiare dell'infanzia e la reticolosi midollare istiocitica dell'adulto appare infatti notevole, tanto che qualcuno ha prospettato l'ipotesi che i due disordini costituiscano variazioni, in rapporto all'età, di un'unica malattia (v. Warnke e altri, 1975).
Clinicamente la reticolosi eritrofagocitica familiare è contraddistinta da febbre, diminuzione del peso corporeo, epatosplenomegalia talora con ittero, pancitopenia con eventuale sindrome emorragica, coinvolgimento di vari organi e distretti (infiltrati polmonari, manifestazioni neurologiche, ecc.). L'esame del midollo osseo rivela un aumento dei macrofagi, che mostrano fenomeni di emofagocitosi, e dei linfociti, spesso accompagnato da iperplasia eritroblastica (v. Ambruso e altri, 1980).
Il trattamento della reticolosi eritrofagocitica familiare è stato condotto con vari regimi terapeutici - antibiotici, trasfusionali e citostatici - nessuno dei quali, tuttavia, si è mostrato in grado di influenzarne il decorso rapidamente mortale. Abitualmente il decesso interviene per complicanze emorragiche, infettive o neurologiche.
f) Malattie caratterizzate dalla proliferazione di istiociti ben differenziati.
1. Malattia di Hand-Schüller-Christian. - Questa malattia, che la maggior parte degli autori ritiene essere la forma cronica disseminata dell'istiocitosi X e che alcuni oggi considerano un'entità nosologica separata dalla malattia di Abt-Letterer-Siwe (v. Lieberman e altri, 1969), è sostenuta dalla proliferazione focale, ma pluridistrettuale, di istiociti ben differenziati.
Dal punto di vista anatomopatologico gli organi più frequentemente colpiti sono le ossa piatte (soprattutto il cranio e il bacino), ma accumuli istiocitari si possono osservare anche nelle ossa lunghe, nella pelle e nei polmoni. La caratteristica istologica più saliente è costituita dalla presenza nelle lesioni di ‛cellule xantomatose' o ‛schiumose' (foamy cells degli autori anglosassoni): trattasi di cellule voluminose, mono- o plurinucleate, il cui citoplasma appare chiaro e vacuolato per lo scioglimento delle sostanze grasse contenutevi. Al reperto di questi elementi tipici si accompagna quello di quote variabili di linfociti, di plasmacellule e di granulociti neutrofili ed eosinofili.
Clinicamente la malattia comincia a manifestarsi nei primi quattro anni di vita con la comparsa di rarefazioni ossee craniche, singole o multiple, di dimensioni variabili (da lesioni appena rilevabili a lacune ampie, irregolari, ‛a carta geografica'). Nel 50% dei casi compare diabete insipido per infiltrazione dell'ipofisi, dell'ipotalamo e/o del peduncolo ipofisario. Nel 33% dei casi si manifesta un esoftalmo, mono- o bilaterale, per infiltrazione orbitaria. La classica triade, lesioni ossee - diabete insipido - esoftalmo, non è di riscontro così frequente come si pensava in passato, ma è rilevabile in non più del 10% dei pazienti (v. Avery e altri, 1957). Fra le manifestazioni cliniche comuni nella malattia di Hand-Schüller-Christian segnaleremo i rashes eritematosquamosi talora difficilmente distinguibili da banali dermatiti seborroiche, le otiti croniche associate a lesioni della mastoide, le lesioni polmonari in genere di modesta entità.
L'evoluzione procede lentamente, nell'arco di più anni. In diversi malati (fra il 30 e il 50% dei casi) si osserva la stabilizzazione prolungata o anche la guarigione con lenta risoluzione delle lesioni; in altri sopravviene un aggravamento con comparsa di nuove localizzazioni e di complicazioni infettive e polmonari (v. Avery e altri, 1957; v. Lieberman e altri, 1969).
2. Granuloma eosinofilo dell'osso, unifocale e plurifocale. - La difficoltà che si incontra talora nella diagnosi differenziale istologica e soprattutto clinica tra la malattia di Hand-Schüller-Christian e la forma plurifocale del granuloma eosinofilo dell'osso giustifica l'inclusione di questa affezione fra le malattie del SRE. In essa, infatti, è possibile la comparsa di manifestazioni sistemiche tali da dar luogo a una sintomatologia analoga a quella osservabile nella malattia di Hand-Schüller-Christian.
Il granuloma eosinofilo colpisce più frequentemente il bambino già grandicello e il giovane adulto; può localizzarsi in qualsiasi punto dello scheletro, ma più frequentemente nel cranio, nel bacino, nelle coste e nei femori. La lesione viene per lo più rivelata da dolori ossei, da una tumefazione o da una frattura spontanea o patologica (v. Schaison e Seligman, 1972).
La forma unifocale presenta una prognosi ottima, dal momento che può guarire prontamente in seguito all'asportazione e al trattamento con radiazioni ionizzanti. La prognosi della forma plurifocale comporta invece le stesse riserve della malattia di Hand-Schüller-Christian.
3. Granulomi eosinofili extraossei e istiocitosi focale. - I granulomi eosinofili extraossei colpiscono per lo più il tratto grastroenterico (v. Salmon e Paulley, 1967) e il polmone (v. Hoffman e altri, 1962). I loro rapporti con il granuloma eosinofilo dell'osso e con la patologia neoplastica e infiammatoria sono ben lungi dall'essere chiariti, così che non si è ancora proceduto al loro definitivo inquadramento nosologico.
Le stesse riserve si possono porre per quelle rare forme di istiocitosi focale benigna segnalate da alcuni autori in vari organi e tessuti (v. Cline e Golde, 1973).
6. Appendice: hairy cell leukemia o reticoloendoteliosi leucemica.
In passato si parlava di reticoloendoteliosi leucemica, secondo la denominazione introdotta da Ewald (v., 1923), o di reticulum cell leukemia; successivamente era stata isolata una particolare leucemia a cellule ‛capellute' (hairy cell leukemia o tricoleucemia) che fu considerata da molti autori come l'equivalente della reticoloendoteliosi leucemica, così che da qualche tempo in molte classificazioni delle malattie del SRE non figura più la reticoloendoteliosi leucemica, malgrado la non sicura identificabilità di tutti i casi descritti con la hairy cell leukemia. Anche se l'interpretazione dell'origine reticoloendoteliale delle cellule hairy è attualmente molto controversa, noi riteniamo che non si possa oggi trattare della patologia del sistema reticoloendoteliale senza accennare a questa forma. Se da alcuni autori le hairy cells sono state considerate in passato come cellule reticolari o istiociti (v. Di Guglielmo e Baserga, 1952; v. Bouroncle e altri, 1958; v. Yam e altri, 1968; v. Mitus e altri, 1961; v. Mitus, 1971), da altri esse sono state interpretate come linfociti (v. Duhamel e Guerra, 1966; v. Rubin e altri, 1969), e nemmeno i progressi immunologici e citochimici degli ultimi dieci anni hanno permesso un definitivo chiarimento in proposito. Così, alcuni autori hanno portato un buon numero di prove a favore della natura B-linfocitaria delle hairy cells (v. Catovsky e altri, 1974; v. Debusscher e altri, 1975; v. Golde e altri, 1976 e 1977; v. Salsano e altri, 1979), mentre altri hanno potuto documentare in queste cellule caratteri propri del MPS (v. Jaffè e altri, 1974; v. King e altri, 1975; v. Scheinberg e altri, 1976; v. Seshadri e altri, 1976). Noi riteniamo, in accordo con Grusovin e altri (v., 1977) e con Variakojis e altri (v., 1980), che le hairy cells non possano essere raggruppate in alcuna linea cellulare definita, e che si tratti di ‟elementi con caratteristiche proprie in virtù di una loro particolare specializzazione funzionale o in conseguenza della peculiarità della noxa neoplastica e/o dell'influenza di fattori ambientali" (v. Grusovin e altri, 1977).
Non si può quindi oggi considerare già risolta la nosologia di questa discussa malattia, linfoproliferativa per alcuni, istiocitoproliferativa per altri. Non si deve trascurare, d'altra parte, la possibilità che la reticoloendoteliosi leucemica sia una sindrome che riunisce, sotto un profilo clinico e citologico sovrapponibile, disordini proliferativi di stipiti cellulari diversi.
Dal punto di vista anatomopatologico la hairy cell leukemia è caratterizzata da rilevante splenomegalia (nel 50% dei casi il peso della milza è superiore ai 2.000 g) ed epatomegalia moderata, senza alterazioni linfonodali e cutanee. Gli elementi che infiltrano la milza, il midollo osseo e il fegato hanno un diametro di 10-18 μm, nucleo eccentrico per lo più ovale a cromatina omogenea, meno coartata che nel linfocito e meno lassa che nei classici blasti leucemici; il loro apparato nucleolare è variabile, nel 50% assai evidente, nel rimanente 50% irrilevante. In alcune cellule possono essere osservati i classici villi citoplasmatici, che assumono il significato di carattere patognomonico: questo aspetto spiculato delle hairy cells è massimamente evidente negli strisci di sangue periferico (v. fig. 14) e di midollo osseo e nelle apposizioni spleniche, cosicché la diagnosi di tricoleucemia risulta più facile all'ematologo che all'anatomopatologo, tanto più che la qualità e l'intensità di alcune reazioni citochimiche risultano peculiari delle cellule in questione (v. Grusovin e altri, 1977; v. Variakojis e altri, 1980).
La malattia colpisce con maggiore frequenza soggetti di sesso maschile (con rapporto maschi/femmine pari a 6 : 1) e di media età. Dal punto di vista clinico il rilievo più importante e più frequente è costituito dalla splenomegalia, che si osserva in oltre il 90% dei pazienti. L'epatomegalia è meno frequente, così come la linfoadenomegalia. Dal punto di vista ematologico la massima parte dei pazienti presenta pancitopenia, anche se possono osservarsi piastrinopenia, anemia e leucopenia isolate. Dei 92 casi raccolti in letteratura da Katayama e Finkel (v., 1974), 55 presentavano un numero di globuli bianchi inferiore a 5.000/ml, 28 un numero compreso fra 5.000 e 15.000/ml, e solo 9 più di 15.000 globuli bianchi/ml. Il numero delle hairy cells variava dallo 0 al 99% in singoli pazienti esaminati in momenti diversi e in pazienti diversi. La situazione pancitopenica è sostenuta in gran parte dall'inefficienza del midollo osseo che, nella massima parte dei casi, è quasi compietamente costituito da hairy cells.
La tricoleucemia decorre come malattia cronica, con inizio insidioso e sintomatologia vaga. I pazienti sono spesso capaci di attendere alle loro attività in modo normale, fino alla comparsa di manifestazioni infettive o emorragiche fatali. Circa un terzo dei pazienti viene a morte entro un anno dalla diagnosi, la metà dopo un anno ed entro i 5 anni; gli altri possono sopravvivere oltre i 5 anni, raggiungendo sopravvivenze anche assai notevoli, di oltre 16 anni. La terapia è basata sulla splenectomia, soprattutto in caso di ipersplenismo, e sull'uso di corticosteroidi e citostatici.
Bibliografia.
Ambruso, D. R., Hays, T., Zwarties, W. J., Tubergen, D. G., Favara, B. E., Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213, in ‟Cancer", 190, XLV, pp. 2516-2520.
Andersen, H., Matthiesen, M. E., The histiocyte in human foetal tissues: its morphology, cytochemistry, origin, function, and fate, in ‟Zeitschrift für Zellforschung und mikroskopische Anatomie", 1966, LXXII, pp. 193-211.
Aschoff, L., Ein Beitrag zur Lehre von den Macrophagen auf Grund von Untersuchungen des Herrn Dr. Kiyono, in ‟Verhandlungen der deutschen Gesellschaft für Pathologie", 1913, XVI, p. 107.
Aschoff, L., Das reticulo-endotheliale System, in ‟Ergebnisse der inneren Medizin und Kinderheilkunde", 1924, XXVI, p. 1.
Avery, M. E., McAfee, J. C., Guild, H. G., The course and prognosis of reticuloendotheliosis (eosinophilic granuloma, Schüller-Chritsian disease and Letterer-Siwe disease), in ‟American journal of medicine", 1957, XXII, pp. 636-652.
Barnes, D. W., Loutit, J. F., Haemopoietic stem cells in peripheral blood, in ‟Lancet", 1967, II, pp. 1138-1141.
Baserga, A., Le malattie del sistema reticolo-endoteliale, in ‟Haematologica", 1933, IV, pp. 61-123.
Baserga, A., Generalità e classificazione delle malattie del sistema reticoloistiocitario, in Trattato italiano di medicina interna (a cura di P. Introzzi), parte III, vol. III, Roma 1961, pp. 2015-2018.
Baserga, A., La regolazione della costanza cellulare negli organi emopoietici, in ‟Haematologica", 1961, XLVI, pp. 61-82.
Bertalanffy, F. O., Respiratory tissue: structure, histophysiology, cytodynamics. I. Review and basic cytomorphology. II. New approaches and interpretations, in International review of cytology (a cura di G. H. Bourne e J. F. Danielli), vol. XVI, p. 233, vol. XVII, p. 213, New York 1964.
Bessis, M., Rôle du système réticulo-hiosticytarie, in Cellules du sang normal et pathologique, Paris 1972, pp. 598-599.
Bianchi, C., La funzione emoblastica del sistema istiocitario, Parma 1951.
Bouroncle, B. A., Wiseman, B. K., Doan, C. A., Leukemic reticuloendotheliosis, in ‟Blood", 1958, XIII, pp. 609-630.
Castellino, P., Ferrata, A., Leucemia e morbi affini. Relazione al XXIV congresso della Società Italiana di Medicina Interna, Genova 1914.
Catovsky, D., Galton, D. A. G., Lysozyme activity and nitroblue tetrazolium reduction in leukaemic cells, in ‟Journal of clinical pathology", 1973, XXVI, pp. 60-69.
Catovsky, D., Pettit, J. E., Galton, D. A. G., Spiers, A. S. D., Harrison, C. V., Leukaemic reticuloendotheliosis (hairy cell leukaemia): a distinct clinicopathological entity, in ‟British journal of haematology", 1974, XXVI, pp. 9-27.
Cazal, P., La réticulose hystiomonocytaire, Paris 1946.
Chen, C., Hirsch, J. G., The effects of 2-mercaptoethanol and of peritoneal macrophages on the antibody-forming capacity of non-adherent mouse spleen cell in vitro, in ‟Journal of experimental medicine", 1972, CXXXVI, pp. 604-617.
Cline, M. J., Biochemistry and function of monocytes and macrophages, in Hematology (a cura di W. J. Williams, E. Beutler, A. J. Erslev e R. W. Rundles), New York 19772, pp. 869-874.
Cline, M. J., Golde, D. W., A review and reevaluation of the histiocytic disorders, in ‟American journal of medicine", 1973, LV, pp. 49-60.
Cline, M. J., Golde, D. W., Cellular interactions in haematopoiesis, in ‟Nature", 1979, CCLXXVII, pp. 177-181.
Cline, M. J., Sumner, M. A., Bone marrow macrophage precursors. I. Some functional characteristics of the early cells of the mouse macrophage series, in ‟Blood", 1972, XL, pp. 62-69.
Cohn, Z. A., Endocytosis and intracellular digestion, in Mononuclear phagocytes (a cura di R. van Furth), Oxford 1970, pp. 121-132.
Crosby, W. H., Structure and fucntions of the spleen, in Hematology (a cura di W. J. WIlliams, E. Beutler, A. J. Erslev e R. W. Rundles), New York 19772, pp. 74-81.
Debusscher, L., Bernheim, J. L., Collard-Range, E., Govaerts, A., Hooghe, R., Lejeune, F. J., Zeicher, M., Stryckmans, P. A., Hairy cell leukemia: functional, immunologic, kinetic, and ultrastructural characterization, in ‟Blood", 1975, XLVI, pp. 495-507.
Di Guglielmo, G., La patologia e la clinica del sistema reticolo-endoteliale, in ‟Haematologica", 1926, VII, p. 481.
Di Guglielmo, G., Sul sistema reticolo-endoteliale, in ‟Bollettino della Società italiana di biologia sperimentale", 1926, I, p. 23.
Di Guglielmo, G., Il sistema reticolo-endoteliale, in ‟Bollettino della Società italiana di biologia sperimentale", 1927, II, p. 937.
Di Guglielmo, G., Le malattie eritremiche, in ‟Medicina e biologia", 1943, III, p. 105.
Di Guglielmo, G., Les maladies érythrémiques, in ‟Revue d'hématologie", 1946, I, p. 355.
Di Guglielmo, G., Baserga, A., Pathologie du système réticulo-endothélial, in Encyclopédie médico-chirurgicale, 13034 A10 A30 A70, Paris 1952.
Doede, K. G., Rappaport, H., Long-term survival of patients with acute differentiated histiocytosis (Letterer-Siwe disease), in ‟Cancer", 1967, XX, pp. 1782-1795.
Downey, H., Monocytic leukemia and leukemic reticulo-endotheliosis, in Handbook of hematology (a cura di H. Downey), vol. II, New York 1938, p. 1275.
Duhamel, G., Guerra, L., Une syndrome hématologique difficile à définir: la myélofibrose lymphoïde, in ‟Presse médicale", 1966, LXXIV, pp. 585-590.
Epstein, E., Die generalisierten Affektionen des histiozytären Zellsystems (Histiozytomatosen), in ‟Medizinische Klinik", 1925, XXI, p. 40.
Ewald, O., Die leukämische Reticuloendotheliose, in ‟Deutsches Archiv für klinische Medizin", 1923, CXLII, pp. 222-228.
Farquhar, J. W., Claireaux, A. E., Familial haemophagocytic reticulosis, in ‟Archives of disease in childhood", 1952, XXVII, pp. 519-525.
Ferrata, A., Le emopatie, vol. I, Roma-Milano-Napoli 1933, p. 150.
Fleischmann, P., Der zweite Fall von Monozytenleukämie, in ‟Folia haematologica", 1916, XX, p. 20.
Florey, H., General pathology, London 1962.
Furth, R. van, Origin and kinetics of monocytes and macrophages, in ‟Seminars in hematology", 1970, VII, pp. 125-141.
Furth, R. van, The origin and turnover of promonocytes, monocytes and macrophages in normal mice, in Mononuclear phagocytes (a cura di R. van Furth), Oxford 1970, pp. 151-165.
Furth, R. van (a cura di), Mononuclear phagocytes in immunity, infection and pathology, Oxford 1975.
Furth, R. van, Mononuclear phagocytes: functional aspects, den Haag-Dordrecht 1980.
Furth, R. van, Cohn, Z. A., The origin and kinetics of mononuclear phagocytes, in ‟Journal of experimental medicine", 1968, CXXVIII, pp. 415-435.
Gall, E. A., The cytological identity and interrelation of mesenchymal cells of lymphoid tissue, in ‟Annals of the New York Academy of Sciences", 1958, LXXIII, pp. 120-130.
Gall, E. A., Mallory, T. B., Malignant lymphoma. A clinico-pathologic survey of 618 cases, in ‟American journal of pathology", 1942, XVIII, pp. 381-429.
Gill, P. G., Waller, C. A., Quantitative aspects of human monocyte function and its measurement in cancer patients, in The macrophage and cancer (a cura di K. James, B. McBride e A. Stuart), Edinburgh 1977, pp. 375-385.
Golde, D. W., Quan, S. G., Cline, M. J., Hairy cell leukemia: ‛in vitro' culture studies, in ‟Annals of internal medicine", 1976, LXXXV, pp. 78-79.
Golde, D. W., Stevens, R. H., Quan, S. G., Saxon, A., Immunoglobluin synthesis in hairy cell leukemia, in ‟British journal of haematology", 1977, XXXV, pp. 359-365.
Goud, T. J. L. M., Schotte, C., Furth, R. van, Identification of the monoblast in mononuclear phagocyte colonies grown in vitro', in Mononuclear phagocytes in immunity, infection and pathology (a cura di R. van Furth), Oxford 1975, pp. 189-203.
Grusovin, G. D., Leucemie acute, in Trattato di anatomia e istologia patologica (a cura di G. Lanza), vol. I, Padova 1974, pp. 709-731.
Grusovin, G. D., Castoldi, G., Scapoli, G., Anzanel, D., Osservazioni citomorfologiche e funzionali in sei casi di ‛hairy cell leukemia', in ‟Haematologica", 1977, LXII, pp. 23-48.
Heyden, H. W. van, Die ortständigen Knochenmarkzellen, in Das Knochenmark (a cura di W. Queisser), Stuttgart 1978, pp. 99-107.
Hibbs, J. B. Jr., Taintor, R. R., Chapman, H. A. Jr., Weinberg, J. B., Macrophage tumor killing: influence of the local environment, in ‟Science", 1977, CXCVII, pp. 279-282.
Hoffman, L., Cohn, J. E., Gaensler, E. A., Respiratory abnormalities in eosinophilic granuloma of the lung. Long term study of fives cases, in ‟New England journal of medicine", 1962, CCLXVII, pp. 577-589.
Hopper, D. G., Pimm, M. V., Macrophages vs. cancer, in ‟Lancet", 1976, II, pp. 255-256.
Huhn, D., Meister, P., Malignant histiocytosis. Morphologic and cytochemical findings, in ‟Cancer", 1978, XLII, pp. 1341-1349.
Humphrey, J. H., Summing up. A layman's view, in Mononuclear phagocytes (a cura di R. van Furth), Oxford 1970, pp. 625-634.
Jaffè, E. S., Schevach, E. M., Frank, M. M., Green, I., Leukemic reticuloendotheliosis: presence of a receptor for cytophilic antibody, in ‟American journal of medicine", 1974, LVII, pp. 108-114.
Jaffè, E. S., Schevach, E. M., Sussman, E. H., Frank, M. M., Green, I., Berard, C. W., Membrane receptor sites for the identification of lympho-reticular cells in benign and malignant conditions, in ‟British journal of cancer", 1975, XXXI (suppl. II), pp. 107-120.
Jaffè, R. H., The reticulo-endothelial system, in Handbook of hematology (a cura di H. Downey), vol. II, New York 1938, p. 973.
Jones, T. C., Attachment and ingestion phases of phagocytosis, in Monuclear phagocytes in immunity, infection and pathology (a cura di R. van Furth), Oxford 1975, pp. 269-282.
Katayama, I., Finkel, H.E., Leukemic reticuloendotheliosis. A clinicopathologic study with review of the literature, in ‟American journal of medicine", 1975, LVII, pp. 115-126.
Katz, D. H., Lymphocyte differentiation, recognition, and regulation, New York 1977.
Kelemen, E., Calvo, W., Fliedner, T. M., Atlas of human hemopoietic development, Berlin 1979.
King, G. W., Hurtubise, P. E., Sagone, A. L., LoBuglio, A. F., Metz, E. N., Leukemic reticuloendotheliosis. A study of the origin of the malignant cell, in ‟American journal of medicine", 1975, LIX, pp. 411-416.
Kiyono, K., Die vitale Karminspeicherung. Ein Beitrag zur Lehre von der vitalen Färbung mit besonderer Berücksichtigung der Zelldifferenzierung im entzündeten Gewebe, Jena 1914.
Kiyono, K., Zur Frage der histiozytären Blutzellen, in ‟Folia haematologica", 1914, XVIII, p. 149.
Kleihauer, E., Hämatologie, Berlin 1978.
Koch, A. L., Miller, C., A mechanism for keeping mutations in check, in ‟Journal of theoretical biology", 1965, VIII, pp. 71-80.
Kurland, J. J., Broxmeyer, I. A., Pelus, L. M., Bockman, R. S., Moore, M. A. S., Role for monocyte-macrophage-derived colony-stimulating factor and prostaglandin E in the positive and negative feedback control of myeloid stem cell proliferation, in ‟Blood", 1978, LII, pp. 388-407.
Landau, M., McNee, J. W., Zur Physiologie des Cholesterinstoffwechsels, in ‟Beiträge zur pathologischen Anatomie und zur allgemeinen Pathologie", 1914, LVIII, p. 667.
Lanza, G., Linfonodi, in Trattato di anatomia e istologia patologica (a cura di G.Lanza), vol. I, padova 1974, pp. 781-880.
Lessin, L.S., Bessis, M., Douglas, S. D., Morphology of monocyts and macrophages, in Hematology (a cura di W. J. Williams, E. Beutler, A. J. Erslev e R. W. Rundles), New York 19772, pp. 851-860.
Letterer, E., Aleukämische Retikulose (ein Beitrag zu den proliferativen Erkrankungen des Retikuloendothelialapparates), in ‟Frankfurter Zeitschrift für Pathologie", 1924, XXX, p. 377.
Lichtenstein, L., Histiocytosis X. Integration of eosinophilic granuloma of bone, ‛Letterer-Siwe disease', and ‛Schüller-Christian disease' as related manifestations of a single nosologic entity, in ‟Archives of pathology", 1953, LVI, pp. 84-102.
Lieberman, P. H., Jones, C. R., Dargeon, H. W. K., Bagg, C.F., A reppraisal of eosinophilic granuloma of bone, Hand-Schüller-Christian syndrome, and Letterer-Siwe syndrome, in ‟Medicine", 1969, XLVIII, pp. 375-400.
Loutit, J. F., Biocycles in the reticulo-endothelial system, in ‟Annals of the New York Academy of Sciences", 1960, LXXXVIII, pp. 122-133.
Lukes, R. J., Collins, R. D., Immunologic characterization of human malignant lymphomas, in ‟Cancer", 1974, XXXIV, pp. 1488-1503.
Lukes, R. J., Parker, J. W., Taylor, C. R., Tindle, B. H., Cramer, A. D., Lincoln, T. L., Immunologic approach to non-Hodgkin's lymphoms and related leukemias. Analysis of the results of multiparameter studies of 425 cases, in ‟Seminars in hematology", 1978, XV, pp. 322-351.
Mackaness, G. B., The immunological basis of acquired cellular resistance, in ‟Journal of experimental medicine", 1964, CXX, pp. 105-120.
Mecnikov, I. I., Lectures on the comparative pathology of inflammation, London 1893.
Mecnikov, I. I., Immunity in infective diseases, London 1905.
Mendelsohn, G., Eggleston, J. C., Mann, R. B., Relationship of lysozyme (muramidase) to hiostiocytic differentiation in malignant histiocytosis. An immunohistochemical study, in ‟Cancer", 1980, XLV, pp. 273-279.
Meuret, G., Monozytopoese beim Menschem, München 1974.
Meuret, G., Disorders of the mononuclear phagocyte system. An analytical review, in ‟Blut", 1977, XXXIV, pp. 317-328.
Meuret, G., Hoffmann, G., Monocyte kinetic studies in normal and disease states, in ‟British journal of haematology", 1973, XXIV, pp. 275-285.
Mitus, W. J., Hairy cells and isoenzymes, in ‟New England journal of medicine", 1971, CCLXXXIV, pp. 389 ss.
Mitus, W. J., Mednicoff, I. B., Wittels, B., Dameshek, W., Neoplastic lymphoid reticulum cells in the peripheral blood: a histochemical study, in ‟Blood", 1961, XVII, pp. 206-215.
Mosier, D. E., Pierce, C. W., Functional maturation of thymic lymphocyte populations in vitro, in ‟Journal of experimental medicine", 1972, CXXXVI, pp. 1484-1500.
Nathan, C. F., Murray, H. W., Cohn, Z. A., The macrophage as an effector cell, in ‟New England journal of medicine", 1980, CCCIII, pp. 622-626.
Nathwani, B. N., Kim, H., Rappaport, H., Solomon, J., Fox, M., Non-Hodgkin's lymphomas, in ‟Cancer", 1978, XLI, pp. 303-325.
Nelson, D. S., Endocytosis by macrophages, in Macrophages and immunity (a cura di D. S. Nelson), Amsterdam 1969, p. 94.
Nelson, D. S., Immunobiology of the macrophage, New York 1976.
North, R. J., The mitotic potential of fixed phagocytes in the liver as revealed during the development of cellular immunity, in ‟Journal of experimental medicine", 1969, CXXX, pp. 315-326.
North, R. J., L'endocitosi, in ‟Aggiornamenti in ematologia", 1970, VII, pp. 216-230.
??? Opsonins, in ‟New England journal of medicine", 1970, CCLXXXII, pp. 391-392.
Osgood, E. E., Number and distribution of human hemic cells, in ‟Blood", 1954, IX, pp. 1141-1154.
Peschle, C., Erythropoiesis, in ‟Annual review of medicine", 1980, XXI, pp. 303-314.
Pittaluga, G., Las enfermedades del sistema reticulo-endothelial, Madrid 1935.
Rabinovitch, M., Phagocytic recognition, in Mononuclear phagocytes (a cura di R. van Furth), Oxford 1970, pp. 299-313.
Rappaport, H., Hematopoietic system, Washington 1966, pp. 91-206.
Rappaport, H., Winter, W. J., Hicks, E. B., Follicular lymphoma. A reevaluation of its position in the scheme of malignant lymphoma, based on a survey of 253 cases, in ‟Cancer", 1956, IX, pp. 792-821.
Reschad, H., Schilling-Torgau, V., Über eine neue Leukämie durch echte Übergangsformen (Splenozyten-Leukämie) und ihre Bedeutung für die Selsbtändigkeit dieser Zellen, in ‟Münchener medizinische Wochenschrift", 1913, LX, pp. 1981-1984.
Robb-Smith, A. H.T., Reticulosis and reticulosarcoma: a histological classification, in ‟Journal of pathology and bacteriology", 1938, XLVII, pp. 457-480.
Rosenthal, A. S., Lipsky, P. E., Schevach, E. M., Macrophagelymphocyte interaction: morphologic and functional correlates, in Mononuclear phagocytes in immunity, infection and pathology (a cura di R. van Furth), Oxford 1975, pp. 813-825.
Roser, B., The distribution of intravenous injected peritoneal macrophages in the mouse, in ‟Australian journal of experimental biology and medical science", 1965, XLIII, pp. 553-562.
Roser, B., The migration of macrophages in vivo, in Mononuclear phagocytes (a cura di R. van Furth), Oxford 1970, pp. 166-174.
Rubin, A. D., Douglas, S. D., Chessin, L. N., Glade, P. R., Dameshek, W., Chronic reticulo-lymphocytic leukemia. Reclassification of ‛leukemic reticuloendotheliosis' through functional characterization of the circulating mononuclear cells, in ‟American journal of medicine", 1969, XLVII, pp. 149-162.
Russell, S. W., McIntosh, A. T., Macrophages isolated from regressing Moloney sarcomas are more cytotoxic than those recovered from progressing sarcomas, in ‟Nature", 1977, CCLXVIII, pp. 69-71.
Saba, T. M., Physiology and physiopathology of the reticuloendothelial system, in ‟Archives of internal medicine", 1970, CXXVI, pp. 1031-1052.
Salmon, P. R., Paulley, J. W., Eosinophilic granuloma of the gstrointestinal tract, in ‟Gut", 1967, VIII, pp. 8-14.
Salsano, F., Pisarri-Salsano, S., Ciancarelli, M. P., Piantelli, M., Laurioli, L., Musiani, P., Structural and functional characteristics of hairy cells, in ‟Acta haematologica", 1979, LXI, pp. 184-193.
Schaison, G., Seligman, M., Istiocitosi X, in Ematologia pediatrica (a cura di C. Alagille, J. Chaissegneaux, J. Dausset, J. Darmont, G. Meshaka e M. Seligman), Roma 1972, pp. 429-448.
Scheinberg, M., Brenner, A. I., Sullivan, A. L., Cathcart, E. S., Katayama, I., The heterogeneity of leukemic reticuloendotheliosis, ‛hairy cell leukemia'. Evidence for its monocytic origin, in ‟Cancer", 1976, XXXVII, pp. 1302-1307.
Schittenhelm, A., Die Klinik des reticuloendothelialen Systems, in Handbuch der Krankheiten der blutbildenden Organe (a cura di A. Bethe, G. von Bergmann, G. Embden e A. Ellinger), vol. II, Berlin 1925, p. 492.
Seshadri, R. S., Brown, E. J., ZIpursky, A., Leukemic reticuloendotheliosis. A failure of monocyte production, in ‟New England journal of medicine", 1976, CCXCV, pp. 181-184.
Shima, K., Dannenberg, A. M., Ando, M., Chandrasekhar, S., Seluzicka, J. A., Fabrikant, J. I., Macrophage accumulation, division, maturation and digestive and microbicidal capacities in tuberculous lesions. I. Studies involving their incorporation of triated thymidine and their content of lysosomal enzymes and bacilli, in ‟American journal of pathology", 1972, LXVII, pp. 159-180.
Skarin, A. T., Canellos, G. P., Chemioterapia dei linfomi non-Hodgkin avanzati, in ‟Ematologia clinica", 1980, IX, pp. 151-168.
Snyderman, R., Meadows, L., Holder, W., Wells, S. Jr., Abnormal monocyte chemotaxis in patients with breast cancer: evidence for a tumor-mediated effect, in ‟Journal of the National Cancer Institute", 1978, LX, pp. 737-740.
Spector, W. G., Mariano, M., Macrophage behaviour in experimental granulomas, in Mononuclear phagocytes in immunity, infection and pathology (a cura di R. van Furth), Oxford 1975, pp. 927-942.
Unanue, E. R., Cooperation between mononuclear phagocytes and lymphocytes in immunity, in ‟New England journal of medicine", 1980, CCCIII, pp. 977-985.
Unanue, E. R., Cerottini, J. C., La funzione dei macrofagi nella risposta immunitaria, in ‟Aggiornamenti in ematologia", 1970, VII, pp. 302-333.
Variakojis, D., Vardiman, J. W., Golomb, H. M., Cytochemistry of hairy cells, in ‟Cancer", 1980, XLV, pp. 72-77.
Volterra, M., Ricerche sul sistema reticoloistiocitario, in ‟Lo sperimentale. Archivio di biologia normale e patologica", 1927, LXXXI, p. 319.
Warnke, R. A., Kim, H., Dorfman, R. F., Malignant histiocytosis (histiocytic medullary reticulosis), in ‟Cancer", 1975, XXXV, pp. 215-230.
Whitelaw, D. M., Batho, H. F., Kinetics of monocytes, in Mononuclear phagocytes in immunity, infection and pathology (a cura di R. van Furth), Oxford 1975, pp. 175-188.
Wintrobe, M. M., Ematologia clinica, vol. II, Padova 1973, p. 951.
Wood, G. W., Gollahon, K. A., Detection and quantitation of macrophage infiltration into primary human tumors with the use of cell-surface markers, in ‟Journal of the National Cancer Institute", 1977, LIX, pp. 1081-1087.
Yam, L. T., Castoldi, G. L., Garvey, M. D., Mitus, W. J., Functional cytogenetic and cytochemical study of the leukemic reticulum cells, in ‟Blood", 1968, XXXII, pp. 90-101.
Zoia, L., Sulla clinica del reticoloendotelio, in ‟Atti della Società lombarda di scienze mediche e biologiche", 1925, XV, p. 3.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


