Trasporto assonale
Dizionario di Medicina (2010)
trasporto assonale
Il trasporto intracellulare di proteine e organelli è fondamentale per la sopravvivenza delle cellule ma, dal momento che i neuroni sono cellule estremamente polarizzate, nel sistema nervoso tale processo assume caratteristiche peculiari. I neuroni infatti hanno un corpo cellulare che si prolunga con assoni e dendriti, e spesso tali strutture proiettano a considerevole distanza l’uno dagli altri (nell’uomo, l’assone di un motoneurone può superare anche la lunghezza di 1 m). Infine, anche all’interno di uno stesso assone, i differenti componenti cellulari da trasportare (cargoes) devono essere spostati verso specifici compartimenti subcellulari, come le proteine canale per il sodio che si addensano nei nodi di Ranvier e le proteine sinaptiche che invece sono spedite verso i terminali periferici. Il trasporto assonale può essere distinto a seconda della direzione in anterogrado o retrogrado, e in base alla velocità in lento o veloce. Gli spostamenti dei materiali da trasportare lungo l’assone sono attuati grazie a specifici motori molecolari. [➔ geni omeotici e cervello; invecchiamento cerebrale; neurodegenerazione; neurone; sinapsi] Storicamente i primi esperimenti finalizzati allo studio del t. a. furono eseguiti nel 1948 da Paul A. Weiss che osservo come, in conseguenza della legatura del nervo sciatico, il citoplasma che decorre all’interno della fibra nervosa (assoplasma) tende ad accumularsi a monte della legatura. Weiss concluse pertanto che l’assoplasma si muove in senso distale a velocità lenta e costante dal corpo cellulare verso le terminazioni, un processo a cui dette il nome di flusso assoplasmatico. Accanto all’esame microscopico diretto degli assoni in coltura, iniziato nel 1920, le ricerche svolte in numerosi laboratori verso la meta degli anni Sessanta dimostrarono che esiste, in aggiunta al movimento lento della massa assoplasmatica, una seconda componente di trasporto cineticamente più rapida. Il movimento di strutture subcellulari nei processi nervosi venne infatti studiato tramite l’uso di traccianti ‘caldi’, cioè marcando le proteine sintetizzate nei corpi cellulari dei gangli delle radici dorsali per iniezione di amminoacidi radioattivi. Successivamente ai primi studi condotti da Bernice Grafstein nei neuroni ottici del pesce rosso (Carassius auratus), gli studiosi Sidney Ochs e Raymond J. Lasek iniettarono un amminoacido marcato in un ganglio spinale di gatto e, dopo aver sacrificato l’animale, a intervalli di tempo regolari recisero il nervo sciatico in segmenti consecutivi cosi da misurare la distribuzione di radioattività. L’entità del trasporto fu quindi misurata dalla radioattività presente nei segmenti consecutivi di fibre di lunghezza uniforme recisi lungo il decorso del nervo stesso, mentre l’andamento del trasporto fu espresso dalla distribuzione delle proteine marcate, in funzione della distanza lungo l’assone e del tempo di iniezione. Grazie a tali studi si comprese che il t. a. ha le seguenti caratteristiche: e sensibile agli inibitori della sintesi proteica; e indipendente dal corpo cellulare, in quanto ha luogo anche in fibre nervose separate dal soma; dipende in maniera critica dal metabolismo ossidativo. In seguito, e grazie allo sviluppo delle tecniche microscopiche che accoppiano la luce all’uso di telecamere, David S. Forman e i suoi colleghi misero in evidenza che il movimento di cargoes di grandi dimensioni (probabilmente mitocondri) e di tipo saltatorio, cioè procede a scatti in direzione sia anterograda sia retrograda (da e verso il corpo cellulare).
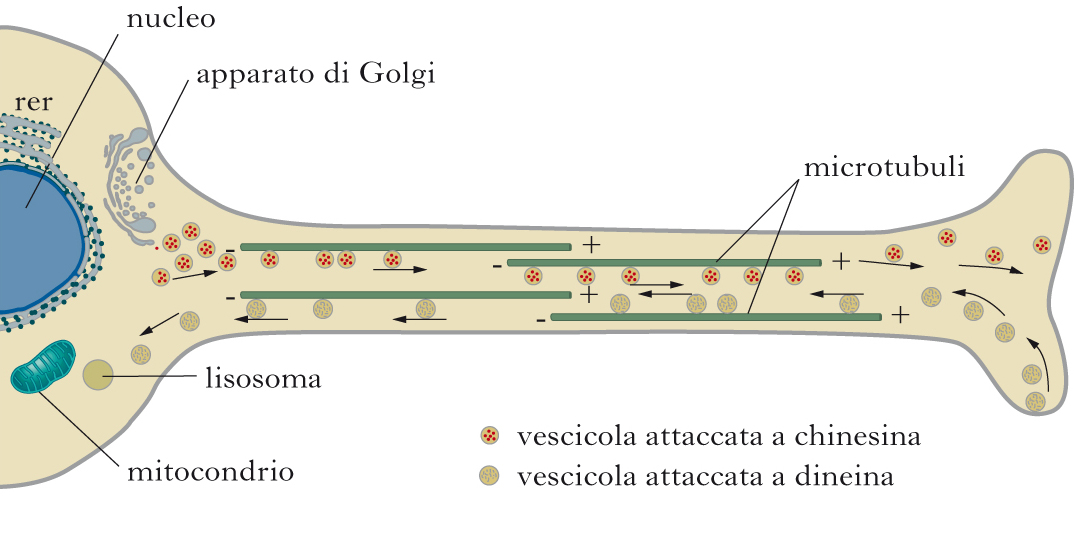
Trasporto anterogrado e retrogrado, microtubuli e motori molecolari
Classicamente vengono distinti due tipi di trasporto lungo l’assone, che si diversificano per velocità e tipo di cargo. Il trasporto in senso anterogrado, cioè dal corpo cellulare verso le terminazioni assoniche, viene utilizzato per il trasporto di tutti i componenti membranosi di nuova sintesi che devono raggiungere le terminazioni nervose; il trasporto retrogrado, in direzione opposta, e invece finalizzato al riciclo degli stessi. Il traffico si svolge poi a due diverse velocità (rapido o lento). Il t. a. lento veicola gli elementi dal pirenoforo all’assone a una velocità di ca. 0,2÷2,5 mm al giorno e pertanto riguarda prevalentemente i costituenti citoscheletrici e altre componenti cellulari stabili, di lunga emivita. Il t. a. veloce, al contrario, interessa soprattutto le vescicole secretorie, gli enzimi del metabolismo dei neurotrasmettitori e i mitocondri, che procedono verso il bottone sinaptico a velocità comprese tra i 50 e i 400 mm al giorno. I maggiori componenti dell’apparato di trasporto neuronale a lunga distanza sono i microtubuli, come dimostrato dalla estrema sensibilità del traffico assonale alla colchicina e alla vinblastina (due alcaloidi capaci di interferire con la struttura di tali componenti del citoscheletro), e i motori molecolari costituiti da chinesina e dineina, o ATPasi microtubuli-associate, capaci di utilizzare l’energia fornita dall’idrolisi dell’ATP per muovere i cargoes lungo i microtubuli stessi. Orientamento e dinamismo dei microtubuli. I microtubuli, insieme all’actina e ai filamenti intermedi, sono una delle componenti strutturali del citoscheletro neuronale. Essi formano una rete citoplasmatica a tubuli polimerici di eterodimeri proteici di α- e β-tubulina. A seguito della disposizione polarizzata delle subunità di tubulina, i microtubuli hanno un orientamento intrinseco in quanto tutti i monomeri α o β di tubulina sono rivolti verso la stessa estremità del microtubulo, determinandone cosi una diversità chimica strutturale. In particolare la β-tubulina e esposta all’estremità plus (diretta verso le sinapsi), mentre la α-tubulina e esposta all’estremità Linus (diretta verso il corpo cellulare). Inoltre, le estremità dei microtubuli differiscono anche cineticamente, in quanto l’estremità detta plus cresce e si accorcia più rapidamente di quella opposta (minus), ed esibisce rispetto a quest’ultima un più robusto dinamismo. Nel corpo cellulare neuronale, come in altri tipi cellulari, la polimerizzazione dei microtubuli avviene attraverso un meccanismo di nucleazione-elongazione, in cui a una lenta fase di formazione di un germe di microtubuli segue una più rapida fase di elongazione a entrambe le estremità per aggiunta, reversibile e non covalente, di dimeri di α- e β-tubulina. La nucleazione avviene all’estremità minus dei microtubuli, in compartimenti subcellulari indicati come centri organizzatori dei microtubuli, o centrosomi, a partire da monomeri di α- o β-tubulina in una conformazione contenente solo GDP (guanosindifosfato) al sito di interscambio, a causa dell’idrolisi del GTP (guanosintrifosfato). Il nucleotide GTP lega l’α-tubulina solubile non polimerizzata a un sito di scambio con la subunità β, ed e successivamente idrolizzato a GDP e Pi (gruppo fosfato), fornendo cosi l’energia chimica necessaria perche la tubulina polimerizzi alle estremità crescenti dei microtubuli. I microtubuli presentano due unici comportamenti di equilibrio non dinamico. Il primo, detto treadmilling, e caratterizzato dalla crescita netta all’estremità plus del microtubulo e dal netto accorciamento all’estremità minus, mentre il secondo, detto instabilità dinamica, e caratterizzato invece dall’alternanza di fasi di crescita sostenuta e di relativamente rapido accorciamento. E interessante riportare che il treadmilling e l’instabilità dinamica non sono comportamenti che si escludono mutuamente, cosicché una specifica popolazione di microtubuli può esibire prima un comportamento e poi l’altro, o entrambi. I parametri che controllano il grado con cui una popolazione di microtubuli esiste sono la composizione isotipica della tubulina, l’entità delle modificazioni postraduzionali e la presenza di proteine regolatorie accessorie. Chinesine e dineine. Dal momento che negli assoni l’orientamento dei microtubuli non e uniforme con le estremità ‘a rapida crescita’ o plus, che puntano verso le sinapsi, e le estremità ‘a lenta crescita’ o minus, che proiettano verso il corpo cellulare, i motori molecolari della famiglia delle chinesine si spostano per lo più unidirezionalmente verso le estremità plus (senso anterogrado) mentre quelli della famiglia delle dineine si muovono in direzione opposta (senso retrogrado) verso l’estremità minus. Le chinesine sono grandi proteine tetrameriche, composte cioè da due catene o subunità pesanti (KHC, Kinesin Heavy Chain) e due catene leggere (KLC, Kinesin Light Chain). La subunità pesante e costituita da due parti: il dominio motore catalitico, che lega i microtubuli, ha attività proteica e conferisce la processi vita e la direzionalità del movimento; la coda, che unitamente alla subunità leggera lega i cargoes e regola l’attività motoria. Mentre il dominio globulare o motore esibisce un alto grado di omologia proteica, l’alta divergenza nella sequenza amminoacidica del dominio che non lega i microtubuli, ma protrude da essi, garantisce invece la specificità diretta del trasporto, che si completa anche, indirettamente, tramite differenti adattatori molecolari che selettivamente riconoscono e legano i diversi cargoes. Le dineine sono complessi proteici multisubunità che contengono due catene o subunità pesanti, le quali a loro volta sono associate a catene o subunità intermedie, leggere-intermedie e leggere. Nelle subunità pesanti che legano i microtubuli risiede l’attività ATPasica, mentre le subunità leggere di più ridotte dimensioni − in partic., p150Glued, dinamitina e dinactina − garantiscono la processi vita del trasporto e controllano il legame dei cargoes. Diversamente dagli assoni, nei dendriti i microtubuli hanno un orientamento misto. Inoltre, come nei bottoni distali assonali, i dendriti terminali contengono pochissimi microtubuli mentre le spine ne sono completamente prive. Cosi, mentre le chinesine e le dineine mediano il trasporto sui microtubuli dal soma lungo l’assone, un altro motore molecolare, la proteina miosina, garantisce il trasporto intraneuronale a breve distanza fino ai terminali e ai coni di crescita, muovendo lungo la rete citoscheletrica dei filamenti di F-actina. La tipica struttura della miosina consiste essenzialmente in due domini, la testa e la coda. La testa, o dominio globulare, è deputata al legame con i filamenti di actina ed è il sito di idrolisi dell’ATP che ne permette il movimento, generalmente verso l’estremità plus, tramite cambiamenti conformazionali dovuti alla diversa affinità della proteina stessa per le molecole da legare (ATP, actina, ADP in ordine decrescente di affinità). La coda è un dominio di struttura allungata che generalmente media le interazioni con molecole trasportatrici e con le altre subunità della proteina stessa.
Selettività del cargo e direzionalità del trasporto intraneuronale
Il trasporto intraneuronale dei cargoes richiede inizialmente il riconoscimento degli stessi da parte dei relativi motori molecolari, un processo selettivo che implica un legame specifico diretto o indiretto − mediato cioè da diverse proteine adattatrici o linker − fra il cargo stesso e il motore proteico. Le differenti e molteplici interazioni tra i numerosi motori proteici e le relative molecole adattatrici basate sul riconoscimento di specifiche sequenze amminoacidiche forniscono così la possibilità di regolare all’interno del neurone con estrema precisione la selettività dei diversi cargoes. La direzionalità del trasporto intraneuronale, cioè il problema di come, una volta scelto il cargo, i motori molecolari si orientano verso gli assoni o i dendriti, è stata a lungo studiata, nonostante le notevoli difficoltà sperimentali riscontrate per la diversa architettura dei processi neuronali. Infatti, mentre gli assoni hanno una lunga traiettoria e tendono a decorrere insieme in fasci, il che facilita l’analisi tramite l’utilizzo di traccianti radioattivi o la tecnica di ligazione del nervo, i dendriti hanno invece una traiettoria più breve e non formano fasci. Considerando dunque che i cargoes sono trasportati non selettivamente verso gli assoni e dendriti, gli studiosi hanno proposto almeno due meccanismi, che non si escludono a vicenda, per la polarità del trasporto. Accanto a un meccanismo di trasporto selettivo − basato cioè su specifiche sequenze amminoacidiche segnale che diversificano i motori a preferenza assonale da quelli dendritici − è stato supposto anche un meccanismo di ritenzione selettiva, che prevede appunto la ritenzione del cargo in un dato compartimento cellulare, o la sua eliminazione da esso nel caso di inappropriata destinazione, rispettivamente per endocitosi inibita o indotta. In aggiunta, come evidenziato da studi strutturali di cristallografia a raggi X, il legame del cargo al motore ne induce un cambio conformazionale che può influenzare la direzionalità del trasporto. Infine, un ulteriore regolazione del trasporto neuronale si realizza a seguito dell’instabilità del macchinario proteico stesso, in quanto la struttura primaria degli adattatori molecolari esibisce numerosi residui amminoacidici di lisina e serina i quali, a seguito di specifiche modificazioni postraduzionali (fosforilazione, ubiquitinazione, sumoilazione e acetilazione), ne influenzano la degradazione e l’accoppiamento tra motore e cargo.
Difetti del trasporto assonale e malattie a carattere neurodegenerativo
L’importanza di un corretto t. a. per la fisiologica funzione e sopravvivenza neuronale è dimostrata dal fatto che i difetti del traffico mediato dai microtubuli contribuiscono nell’uomo alla patogenesi di vari disordini neurodegenerativi, tutti caratterizzati appunto da un aberrante accumulo di organelli e proteine nel corpo cellulare e nell’assone. Per es., la proteina tau è presente nei grovigli neurofibrillari o PHFs (Paired Helical Filaments) dei malati di Alzheimer (AD), la proteina α-sinucleina è il principale costituente dei corpi di Lewy nei pazienti con malattia di Parkinson (PD), mentre i neurofilamenti si accumulano nei soggetti affetti da sclerosi laterale amiotrofica (SLA). Infine, per tutti questi disordini neurologici, è stata riportata una marcata dilatazione assonale caratterizzata da evidenti varicosità, indice appunto di accumulo patogenetico di organelli a seguito di un danno al sistema di traffico neuronale. Diverse sono le possibili cause ipotizzate: • danno ai motori molecolari, chinesina e dineina, come dimostrato dall’osservazione che mutazioni nella subunità della dinactina p150Glued sono geneticamente associate alla disfunzione motoria tipica della SLA; • danno ai cargoes, come per es. accade nei casi familiari di SLA per la proteina mutante SOD-1 (Cu/Zn/superossidodismutasi-1) che attiva alcune vie di trasduzione intracellulari culminanti nell’alterata fosforilazione dei cargoes (per es., neurofilamenti), disturbandone così l’interazione con i relativi motori proteici associati ai microtubuli; • danno ai microtubuli, come accade nell’Alzheimer per la proteina tau la quale, se patologicamente iperfosforilata, perde la sua affinità per i microtubuli causandone la destabilizzazione e il conseguente collasso dell’intera rete citoscheletrica; • danno ai mitocondri, che provoca un ridotto apporto energetico di ATP, come accade in seguito a mutazioni di alcuni geni codificanti le proteine necessarie al mantenimento della bioenergetica mitocondriale e alle difese antiossidanti tramite l’eliminazione dei dannosi radicali liberi; mutazioni di questo tipo sono state individuate in forme familiari di malattia di Parkinson (nei geni parkin, PINK1, DJ-1 e LRRK2), di paraplegia spastica ereditaria o HSP (a carico della proteina paraplegina) o della malattia di Charcot-Marie-Tooth (CMT) (nella proteina mitofusina). Sebbene un potenziale legame patogenetico tra i disordini del t. a. e le sopra citate malattie neurodegenerative sia stato largamente dimostrato, rimangono ancora irrisolte numerose e importanti questioni. Per es., i difetti del traffico neuronale sono causa o conseguenza di tali malattie neurologiche, dal momento che nei modelli transgenici di SLA, PD e AD essi sono evidenti molto tempo prima delle tipiche lesioni patologiche? Inoltre, non è ancora chiaro se gli effetti dannosi sul t. a. sono specifici, ossia causano la distruzione di una singola classe di materiale biologico trasportato, o non specifici, colpevoli invece di una riduzione generalizzata o del blocco fisico di vie multiple di trasporto attraverso l’aggregazione di più cargoes trasportati. Infine, è ancora incerto se le mutazioni umane geneticamente associate a tali disordini neurologici rappresentino esclusivamente una perdita delle normali funzioni fisiologiche della proteina (loss of function) o agiscano con l’acquisto di nuove funzioni tossiche (gain of function), risultando così in proteine dannose che titolano o avvelenano i componenti del trasporto assonale. Una più profonda conoscenza dei motori proteici e dei componenti accessori del t. a., nonché dei relativi meccanismi regolatori, ottenuta tramite lo studio genetico e biochimico condotto in sistemi animali modello, può condurre allo sviluppo di nuove e più promettenti terapie per il trattamento di numerosi disordini ereditari umani caratterizzati da aggregazione proteica, stress ossidativo, disfunzione mitocondriale, infiammazione e difetti del traffico intracellulare. Giuseppina Amadoro
© Istituto della Enciclopedia Italiana - Riproduzione riservata


