Gas
Enciclopedia on line
Termine attribuito nel 1620 dal chimico J.B. van Helmont alle sostanze che si trovano allo stato aeriforme e quindi prive di volume proprio. Lo stato gassoso, come ogni altro stato di aggregazione, dipende dalle condizioni di temperatura e pressione, e non è caratteristico di determinate sostanze: dicendo che una sostanza, per es. l’aria, è un g., si vuol solo dire che essa è tale nelle ordinarie condizioni di temperatura e di pressione, variando le quali può invece presentarsi come un liquido o anche come un solido. In verità si riteneva in passato che alcuni g., detti per tal motivo permanenti o incoercibili, non si potessero liquefare. Oggi si sa che la liquefazione è sempre possibile purché il g. sia portato al di sotto di una certa temperatura critica, caratteristica per ogni g., e venga poi sufficientemente compresso; la sola compressione non basta se il g. si trova a temperatura superiore a quella critica, che per taluni di essi è estremamente bassa (per l’elio, per es., è di −267,8 °C).
Chimica, fisica e tecnica
G. perfetti
Si possono definire g. perfetti i g. che obbediscono esattamente alla legge di Boyle e Mariotte: «a una data temperatura la pressione p di una data massa di g. è inversamente proporzionale al volume V che essa occupa», cioè:
[1] pV = costante.
Per una data massa di un g. perfetto a una data pressione vale la prima legge di Volta e Gay-Lussac, la quale si esprime nella relazione
[2] V = V0 (1 + αt),
dove V0 è il volume del g. alla temperatura di 0 °C, V quello alla temperatura t, α è il cosiddetto coefficiente di dilatazione termica, che ha per tutti i g. perfetti il medesimo valore, pari a 1/273,15 °C−1. A un dato volume vale una legge analoga (seconda legge di Volta e Gay-Lussac, o legge di Charles):
[3] p = p0 (1 + αt),

dove p0 e p sono le pressioni rispettivamen;te alla temperatura di 0 °C e alla temperatura t, e α è un coefficiente che risulta uguale al coefficiente di dilatazione termica sopra definito. Le [1], [2] e [3] costituiscono le relazioni tra pressione, volume e temperatura che, rispettivamente, caratterizzano le trasformazioni reversibili isoterme, isobare, isocore di un g. perfetto. Le trasformazioni adiabatiche reversibili di un g. perfetto sono caratterizzate dalla relazione pVγ=cost, dove γ rappresenta il rapporto tra i calori specifici del g. a pressione e a volume costante. Qualunque sia la trasformazione mediante la quale si fa passare un g. perfetto da uno stato iniziale caratterizzato dal volume V0, dalla pressione p0 e dalla temperatura di 0 °C, a uno stato finale caratterizzato da valori V, p, t del volume, della pressione, della temperatura centigrada, si riconosce che vale la relazione pV=p0V0 (1+αt), detta equazione di stato, o equazione caratteristica dei g. perfetti, la quale può essere essa stessa assunta a definire i g. perfetti. Facendo intervenire in luogo della temperatura centigrada t la temperatura termodinamica T definita dalla relazione T=t+1/α=t+273,15, l’equazione precedente si trasforma nell’altra equivalente pV=p0V0αT, dove la costante p0V0α dipende soltanto dalla quantità del g. considerato (v. l’andamento qualitativo in fig. 1). Precisamente, se ci si riferisce a una mole di un g. qualunque, che occupa, alla pressione di 101.325 Pa (1 atm), e alla temperatura di 273,15 K (0 °C), un volume di 22,414∙10–3m3, l’equazione di stato dei g. perfetti si scrive:
pV=RT,
essendo R, la cosiddetta costante dei g., pari a
R=8,314 joule/K∙mol (1,98 cal/K∙mol).
Se si considera una quantità di g. diversa da una mole, l’equazione di stato deve essere scritta
pV=n R T,
dove n=m/M è il numero di moli, essendo m la massa del g. in questione espressa in grammi ed M la massa molecolare del gas.
Lo studio dell’espansione nel vuoto mostra che l’energia interna U di un g. perfetto dipende esclusivamente dalla temperatura. Applicando il 1° principio della termodinamica a trasformazioni isobare e isocore relative all’unità di massa di g. perfetto, si dimostra che il calore specifico a pressione costante, cp, è legato al calore specifico a volume costante, cv, dalla relazione:

D’altra parte, per il calore specifico a volume costante vale la relazione: cv=dU/dT.
Teoria cinetica dei gas
Le varie proprietà dei g. perfetti possono essere desunte in base a considerazioni statistiche riguardo ai moti delle molecole. I concetti fondamentali che sono alla base della teoria cinetica dei g. possono riassumersi nei termini seguenti. Un g. è costituito da un grandissimo numero di molecole in moto incessante; le loro distanze reciproche sono grandi rispetto alle loro dimensioni, cosicché ogni molecola per la maggior parte del tempo non risente alcuna azione sensibile da parte delle altre e solo di tanto in tanto viene a collidere con esse o con le pareti del recipiente. Si ammette che fra un urto e l’altro (alla pressione ordinaria di 1 atmosfera e alla temperatura di 0 °C una molecola subisce circa 5∙109 urti al secondo) le singole molecole si muovano di moto rettilineo uniforme: con ciò si trascura l’azione della gravità, che modifica assai poco il movimento data la grande velocità delle molecole (dell’ordine di 1000 m/s). Il moto di queste è tanto più veloce quanto più è elevata la temperatura del g., all’aumentare della quale è perciò da attendersi che, in un volume costante, aumenti la pressione sulle pareti. La teoria che da tali ipotesi si sviluppa, attraverso convenienti applicazioni dei metodi statistici, ha ricevuto tante e così dirette, variate e precise conferme sperimentali, da non esservi oggi il minimo dubbio sulla attendibilità delle ipotesi suddette.
La teoria cinetica si basa sugli studi di R. Clausius, di J.C. Maxwell, di L. Boltzmann e di J.W. Gibbs. Per un g. costituito da molecole così piccole rispetto alle loro distanze da poterle considerare puntiformi e tali che non si esercitino tra loro forze sensibili a distanza (così precisamente va considerato nello schema particellare quel che comunemente si chiama un g. perfetto) si dimostra che è:
[4] formula,
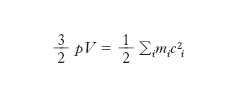
dove mi è la massa della i-esima molecola, ci la sua velocità e la somma è estesa a tutte le molecole del gas. Dunque il prodotto pV è uguale a 2/3 dell’energia cinetica totale delle molecole del g.: se si ammette, come si fa nella teoria cinetica, che questa dipenda soltanto dalla temperatura, se ne deduce che a temperatura costante è pV=cost, cioè si ottiene la legge di Boyle e Mariotte. Se le molecole hanno tutte la stessa massa m e si introduce la loro velocità quadratica media c̅̅2 (definita dall’uguaglianza c̅̅2=(∑c2i)/Ɲ, dove Ɲ è il numero di molecole del g.), la [4] diventa
[5] formula.
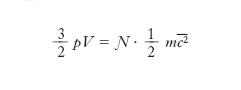
Confrontando la [5] con l’equazione di stato pV=nRT si ottiene (ricordando che n= Ɲ/N, dove N è il numero di Avogadro)
[6] formula,
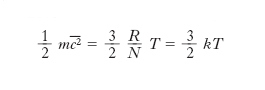
dove k=R/N=1,38∙10–23 J/K è la costante di Boltzmann. Pertanto l’energia cinetica media molecolare mc2/2 risulta proporzionale alla temperatura termodinamica del gas. Tale risultato non è che un caso particolare di risultati più generali che si ottengono come conseguenza della legge di distribuzione di Maxwell-Boltzmann (➔ statistica).
Gas reali
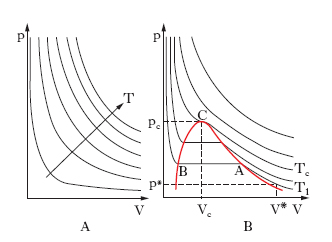
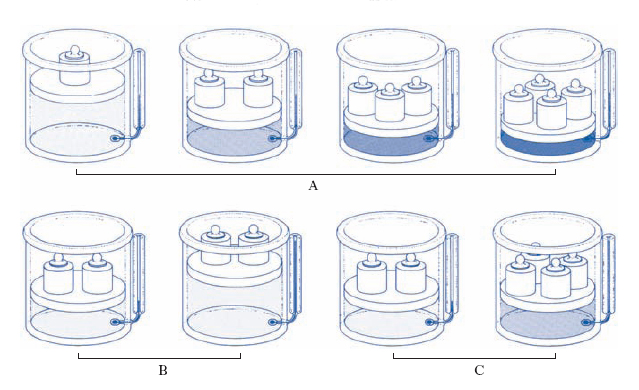
Le leggi cui ubbidiscono i g. reali presentano deviazioni più o meno accentuate da quelle dei g. perfetti: per es., le isoterme non sono più rappresentate, nel piano (p, V), da rami di iperboli equilatere (diagramma di Watt; fig. 2A), come vuole la legge di Boyle per i g. perfetti, ma si presentano come in fig. 2B (diagramma di Andrews). Esse cioè si dividono in due gruppi: quelle relative a temperature inferiori alla temperatura critica, le quali presentano un tratto rettilineo orizzontale, e quelle relative a temperature superiori alla temperatura critica senza il tratto rettilineo orizzontale. L’isoterma Tc corrispondente alla temperatura critica, isoterma critica, presenta un punto di flesso C a tangente orizzontale: C si dice punto critico; la pressione pc e il volume Vc corrispondenti si dicono pressione critica e volume critico. Per quanto riguarda l’andamento delle isoterme al di sotto della temperatura critica, si consideri l’isoterma T1 nella fig. 2A. Assumendo come rappresentativo dello stato iniziale del sistema gassoso il punto di coordinate (p*, V*), si segua l’andamento della compressione isoterma: il volume si riduce all’aumentare della pressione, fino allo stato rappresentato dal punto A; a partire da A, il volume decresce ma la pressione resta costante, in quanto il g. inizia a liquefarsi. La liquefazione dura per tutti gli stati corrispondenti al tratto AB; in B il g. è tutto liquefatto: a causa della molto minore comprimibilità della fase liquida rispetto a quella aeriforme, aumenti relativamente grandi di pressione producono diminuzioni relativamente piccole di volume, cioè l’isoterma è qui assai più inclinata sull’asse dei volumi di quanto non lo fosse nel tratto corrispondente alla fase aeriforme, al di là del ‘punto di rugiada’ A. Come risulta chiaramente dalla fig., il g. non liquefà per sola compressione al disopra della temperatura critica; risulta poi che le isoterme corrispondenti a temperature abbastanza più alte di quella critica quasi coincidono con rami di iperbole equilatera, almeno nel campo delle piccole e medie pressioni: il che giustifica la pratica corrente di assimilare i g. reali a g. perfetti quando essi si trovino ben al di sopra della loro temperatura critica e non siano molto compressi.
Per rappresentare il comportamento dei g. reali sono state proposte diverse equazioni, analoghe alla equazione caratteristica dei g. perfetti, delle quali la più nota è quella (1873) di J.D. van der Waals

,
dove a e b sono due grandezze dipendenti dalla natura del g. in esame e gli altri simboli hanno lo stesso significato che assumono nell’equazione di stato dei g. perfetti; si riconosce che a dà conto delle azioni intermolecolari (nulle in un g. perfetto), mentre b, detto covolume, dà conto del volume proprio delle molecole (assunto nullo per un g. perfetto). Si riportano in tab. le principali caratteristiche di alcuni g. (le densità e i calori specifici s’intendono per temperature prossime a 20 °C).
Gas d’acqua
È uno dei più importanti prodotti della gassificazione, usato come combustibile e come g. di sintesi. Si ottiene insufflando vapor d’acqua su coke rovente; a seconda della temperatura a cui si effettua l’operazione si può avere l’ossidazione del carbonio a ossido o ad anidride carbonica, secondo le reazioni seguenti (in cui si riportano anche le tonalità termiche riferite alla ossidazione di un grammoatomo di carbonio):
[1] C + H2O (vap.) ⇄ CO + H2 − 131 kJ,
[2] C + 2H2O (vap.) ⇄ CO2 + 2H2 − 90 kJ.
La reazione [2] risulta dalla combinazione della [1] con la reazione
[3] CO + H2O (vap.) ⇄ CO2 + H2 + 41 kJ,
che viene chiamata reazione di conversione (o d’equilibrio) del g. d’acqua in quanto tiene conto di tutti i componenti gassosi che prendono parte all’equilibrio. Dalla esotermicità della [3] consegue che quanto più alta è la temperatura a cui avviene la reazione, tanto maggiore è la percentuale di ossido di carbonio presente all’equilibrio nella miscela gassosa; al diminuire della temperatura diminuisce anche la velocità con cui si stabilisce l’equilibrio [3]. Dato che le [1] e [2], cioè le reazioni di formazione del g. d’acqua, sono endotermiche, perché la reazione possa mantenersi occorre fornire al combustibile il calore assorbito nel processo, ciò che si può fare in più modi: a) in modo intermittente; si ricorre a un processo ciclico costituito dall’alternanza di una fase detta di soffio freddo, in cui si insuffla vapore d’acqua producendo g. d’acqua (e raffreddando, di conseguenza, il combustibile rovente), con una fase detta di soffio caldo, in cui si ripristina il livello termico insufflando sul coke aria che ravviva la combustione; naturalmente solo durante la prima fase si produce il g. d’acqua e occorre quindi raccoglierlo separatamente dai g. della fase di combustione; i gassogeni che funzionano secondo questo schema sono forniti di automatismi che permettono di tenere separati i g. prodotti nelle varie fasi; b) in modo continuo; per ottenere ciò è necessario fornire il calore dall’esterno al combustibile (ciò che però si realizza male) o immettere nel gassogeno, insieme al vapore, ossigeno, che fa bruciare una frazione del combustibile: il calore sviluppato mantiene la massa alla temperatura di reazione voluta. Non occorre qui separare i g. dalle due fasi perché, mancando l’azoto dell’aria, i componenti del g. d’acqua non vengono a diluirsi troppo e perché quella parte di anidride carbonica che alla fine è presente nel g. può separarsi facilmente, se necessario; si hanno gassogeni che funzionano su questo principio utilizzando come combustibile sia il coke, sia ligniti, sia carboni fossili minuti.
Il g. d’acqua in genere ha la composizione: idrogeno 48-50%, ossido di carbonio 38-40%, azoto 2-4%, anidride carbonica 4-6%, tracce di metano ecc.; ha un potere calorifico di circa 10.000 kJ/m3; da 1 kg di coke si ha circa 1 m3 di g. d’acqua. G. d’acqua carburato Miscela gassosa combustibile, risultante dall’arricchimento del g. d’acqua con i prodotti gassosi ottenuti per piroscissione di frazioni petrolifere (dai distillati leggeri fino agli oli pesanti).
Gas d’altoforno
G. combustibile, prodotto, durante la preparazione della ghisa all’altoforno, dall’insieme delle reazioni fra il carbonio e gli ossidi di ferro della carica e l’anidride e l’ossi;do di carbonio prodotti durante il processo metallurgico stesso. È costituito da ossido di carbonio (25-30%), da idrogeno e metano (0,5%-3% di ciascuno), da anidride carbonica (10-16%), da azoto (50-60%); ha un potere calorifico di circa 3500-4000 kJ/m3; si ottiene in ragione di circa 4500 m3 per ogni tonnellata di ghisa prodotta e viene utilizzato in parte come g. combustibile negli impianti metallurgici stessi (ricuperatori Cowper), e il rimanente sia per usi termici sia per produzione di energia meccanica o elettrica.
Gas d’aria
Miscela gassosa combustibile, ottenuta facendo passare una corrente d’aria attraverso uno strato sufficientemente spesso di coke incandescente. L’ossigeno dell’aria reagisce con i primi strati del combustibile, i quali bruciando generano anidride carbonica; questa poi reagisce rapidamente con gli strati sovrastanti dando ossido di carbonio, secondo la reazione
CO2+C⇆2CO+172 kJ,
detta reazione d’equilibrio del g. d’aria. Teoricamente il g. d’aria dovrebbe contenere 33,3% di ossido di carbonio e 66,6% di azoto (che è quello che accompagna l’ossigeno nell’aria impiegata nella gassificazione); in pratica, specie se la marcia del gassogeno è sotto i 900 °C, si ha sempre presenza di anidride carbonica e la composizione media del g. d’aria comprende: ossido di carbonio (25-30%), azoto (65-68%); anidride carbonica (2-4%), oltre a piccole quantità di idrogeno, metano, anidride solforosa; il potere calorifico è di circa 4000 kJ/m3; da 1 kg di coke si producono circa 5,6 m3 di g. d’aria.
Gas di città
G. combustibile distribuito nelle abitazioni per usi di riscaldamento (cucine, scaldabagni, stufe ecc.); detto anche g. illuminante (o gas-luce), perché inizialmente (Londra, 1816; in Italia, Torino, 1838) fu usato per l’illuminazione pubblica e privata. Come tale si usava un tempo esclusivamente il g. di distillazione di carboni ad alta temperatura, mentre oggi si usano anche g. di conversione ossidante del g. naturale o di prodotti petroliferi, g. di petrolio liquefatti o miscele di essi con aria e, nei centri abitati prossimi a giacimenti metaniferi o serviti da metanodotti, anche g. naturale: in Italia, anzi, il g. di distillazione di carboni costituisce ormai una piccola parte del g. di città complessivamente distribuito.
Gas di cokeria
Viene ottenuto dalla distillazione dei litantraci eseguita nelle cokerie nelle quali lo scopo principale è la produzione del coke metallurgico; è analogo al g. di distillazione delle officine del g. di città ma si ottiene distillando a temperature più alte, partendo anche da litantraci diversi (con minor contenuto di materie volatili). Si impiega per lo più come g. di riscaldamento nelle industrie metallurgiche.
Gas liquefatti
Miscele di idrocarburi paraffinici (dette impropriamente g. liquidi), per lo più essenzialmente propano e butano con talora piccole percentuali di etano o di idrocarburi non saturi (butilene, etilene ecc.), che alla temperatura normale si lasciano liquefare facilmente (sotto pressione di 2-8 bar) e che, riportati alla pressione atmosferica, passano immediatamente allo stato gassoso. Vengono comunemente indicati con la sigla GPL (G. di Petrolio Liquefatti). Sono ottenuti principalmente dalla distillazione del petrolio grezzo e dal degasolinaggio del g. naturale. Hanno elevato potere calorifico (circa 50.000 kJ/kg) e bassa velocità di propagazione della fiamma; per le loro caratteristiche di combustione richiedono bruciatori particolari, diversi da quelli usati per il comune g. di città. Avendo numero di ottano intorno a 100, sono ottimi carburanti che consentono elevati rapporti di compressione.
I loro impieghi si possono suddividere in tre grandi categorie: come combustibili (per usi domestici: cucina, riscaldamento, talvolta illuminazione; per usi industriali: per la produzione di atmosfere controllate, nella lavorazione dei metalli, per il trattamento termico dei prodotti nelle industrie del vetro e delle ceramiche ecc.); come carburanti; come materia prima per l’industria chimica. I g. liquefatti sono usati anche come componenti del g. di città, sia distribuiti in fase gassosa (il solo propano), attraverso reti proprie di condutture o miscelati con aria (aria propanata), sia dopo trasformazione che, modificandone la natura chimica e le proprietà fisiche, li renda perfettamente intercambiabili con il g. di distillazione del carbone fossile; hanno su quest’ultimo il grande vantaggio di non essere tossici. La loro produzione è cresciuta vertiginosamente dopo la Seconda guerra mondiale, per lo sviluppo dell’industria di raffinazione del petrolio e di degasolinaggio del g. naturale. L’immagazzinamento si effettua in serbatoi fissi, in serbatoi semifissi (carri cisterna e bombole), in serbatoi mobili (autocisterne), in serbatoi a bassa pressione refrigerati, in serbatoi sotterranei (caverne) e in terreno congelato. Il prodotto delle raffinerie viene imbottigliato, in appositi stabilimenti situati possibilmente in prossimità dei centri di consumo, in bombole di varia capacità (5, 10, 15 kg per usi domestici; fino a 100 kg per usi industriali), o trasferito in condotta anche per migliaia di chilometri (USA, Argentina).
Gas naturali
Definizione
I g. naturali si trovano negli strati del sottosuolo e talora ne emanano spontaneamente, ma più spesso sono portati alla luce con trivellazioni anche molto profonde. Per evitare confusioni, si chiama convenzionalmente g. naturale una miscela gassosa di origine naturale costituita essenzialmente da idrocarburi in cui il metano è il componente fondamentale. A esso si associano, anche se in modeste quantità, etano, propano, butano, esano e inoltre anidride carbonica, idrogeno solforato e azoto. In tal modo si distingue questo combustibile, che ha rilevante importanza economica e industriale, dagli altri ‘g. naturali’ – che possono avere anche applicazioni industriali – quali i g. di soffioni e i g. vulcanici, che contengono solo piccole quantità di idrocarburi o non ne contengono affatto. Si distinguono g. naturali umidi e secchi, secondo che insieme al metano siano contenuti o no idrocarburi condensabili, che possono essere recuperati e liquefatti mediante pressione, andando così a costituire quello che commercialmente viene definito g. liquefatto; acidi e dolci, secondo che contengano o no idrogeno solforato o composti organici dello zolfo. Secondo le condizioni del g. nel giacimento, taluni distinguono tra g. non associato, che in giacimento non si trova a contatto con il petrolio o che, pur essendo in contatto, all’erogazione dal pozzo risulta quasi, o totalmente, privo di petrolio; g. associato, a contatto con petrolio, del quale contiene una notevole quantità all’erogazione, e g. in soluzione cioè disciolto, in giacimento, nel petrolio. Infine, vi è il g. disciolto in acqua, come, in Italia, nel Polesine. Vi sono giacimenti enormi di g. naturale non associato, in zone dove non vi è traccia di petrolio (per es.: giacimenti della Valle Padana, alcuni giacimenti russi e giacimenti americani). Durante l’estrazione alcune miscele di g. subiscono il fenomeno della condensazione, che dà origine al cosiddetto g. condensato. Da quest’ultimo, attraverso un processo di degasolinaggio, si ottiene la gasolina naturale. La genesi dei g. associati è strettamente collegata a quella del petrolio; per i g. non associati al petrolio l’ipotesi più attendibile è quella che attribuisce la genesi del g. a materia organica di origine vegetale dispersa nelle rocce sedimentarie, la quale abbia subito processi di fermentazione e carbonizzazione. Le condizioni di giacitura del g. naturale, solo o associato ad altri idrocarburi gassosi o liquidi, sono le stesse di quelle del petrolio.
Raccolta e conservazione
Fra le strutture geologiche favorevoli alla raccolta e conservazione del g. naturale è caratteristica quella ad anticlinale chiusa, o a cupola, costituita da strati porosi protetti superiormente da una copertura impermeabile, in genere argillosa (per es., giacimenti del Lodigiano). Altre giaciture favorevoli possono aversi in corrispondenza di una faglia, che, per es., tronchi lo strato poroso gassifero, tamponandolo lateralmente con strati impermeabili portatisi al contatto dello strato mineralizzato per effetto dello spostamento dovuto alla faglia. Qualora il g. naturale sia associato al petrolio della medesima struttura, esso occupa per la sua minore densità gli orizzonti superiori. La ricerca e la coltivazione dei giacimenti si eseguono con metodi analoghi a quelli usati nei giacimenti petroliferi. Si adotta in genere, nella coltivazione, un maggior distanziamento tra un pozzo e l’altro; inoltre, la pressione del g. nel giacimento assicura sempre l’erogazione del pozzo e non è perciò necessario ricorrere a metodi di recupero secondario come si fa, invece, per il petrolio. Nel caso di g. umidi si ricorre, talvolta, all’iniezione di g. secco in giacimento per evitare che gli idrocarburi liquidi pesanti si condensino nello strato e, quindi, vadano perduti. Le operazioni di produzione vengono generalmente effettuate in cantiere e consistono, di norma: nell’eliminazione dell’acqua (disidratazione) per evitare gli inconvenienti dovuti a condensazione e a formazione di idrati solidi; nella separazione degli idrocarburi superiori liquidi o liquefatti eventualmente presenti (degasolinaggio), suscettibili di impiego a parte; nell’eliminazione di composti solforati (desolforazione), che darebbero altrimenti luogo a inconvenienti in talune utilizzazioni. Talvolta si eliminano e si recuperano l’elio, l’azoto, l’anidride carbonica contenuti nel gas.
Trasporto
Il trasporto del g. naturale avviene sia tramite gasdotto sia, allo stato liquefatto, per nave. La liquefazione del g. naturale consente di risolvere sia il problema dell’immagazzinamento a lungo termine sia quello del trasporto alle grandi distanze, in particolare il trasporto transoceanico, inattuabile con i gasdotti. Gli impianti di liquefazione (➔ criologia) sono normalmente ubicati in corrispondenza delle zone di immagazzinamento o di carico e sono alimentati con tubazioni dai grandi nodi di distribuzione o direttamente dalle aree di estrazione. L’immagazzinamento del g. naturale liquefatto è in genere effettuato alla pressione atmosferica (per eliminare le difficoltà connesse con la costruzione e l’esercizio di recipienti di elevata capacità in grado di resistere a pressione), ma ciò comporta una temperatura di esercizio di circa −160 °C con conseguente necessità di assicurare un sufficiente grado di isolamento termico. Per quanto efficace sia l’isolamento, una certa evaporazione avviene comunque (ogni 24 ore circa 0,1- 0,2% della quantità immagazzinata). Per l’immagazzinamento si impiegano serbatoi metallici in elevazione (a doppia parete con l’intercapedine riempita con materiali isolanti speciali), serbatoi in cemento sia in elevazione sia interrati, caverne naturali o espressamente scavate al di sotto del livello del suolo. Dopo il trasporto con navi metaniere il g. naturale liquefatto viene rievaporato e immesso nella normale rete di distribuzione. Talvolta la rievaporazione viene sfruttata come sorgente di freddo per la conservazione di derrate ecc.
Impiego
Prima della Seconda guerra mondiale, nei paesi che disponevano di risorse sia petrolifere sia di g. naturale (USA, Unione Sovietica ecc.), quest’ultimo era considerato, specie se inquinato da prodotti solforosi, di scarso interesse industriale, tanto che era utilizzato, non sempre razionalmente, unicamente sul posto e cioè nella stessa zona di estrazione. Dopo la Seconda guerra mondiale, invece, il g. naturale è diventato una delle principali fonti di energia e i vantaggi offerti dal suo impiego ne hanno consentito il collocamento, in alcuni settori, anche a prezzi superiori a quelli di altre fonti. In Italia il principale campo d’impiego è rappresentato dalle lavorazioni e tecnologie industriali che consumano circa il 35% del g. naturale; altri campi d’impiego sono dati dal riscaldamento degli ambienti, dagli usi domestici, dagli usi chimici, dalla produzione di energia termoelettrica, dagli impianti con turbomotori a g., dalla trazione. Come combustibile gassoso il g. naturale può essere convenientemente impiegato in quasi tutte le industrie. L’assenza di ceneri e di impurezze ne rendono particolarmente vantaggioso l’impiego nelle industrie del vetro, della ceramica, dei laterizi, dei cementi, nelle industrie metallurgiche (forni di riscaldo, forni Martin-Siemens, nuove tecniche siderurgiche come la riduzione diretta), nell’essiccamento delle vernici e negli impianti di produzione di vapor d’acqua, sia in centrali termiche e termoelettriche sia in varie lavorazioni industriali. L’impiego per usi domestici come g. di città, con qualche modifica ai bruciatori, ha incontrato favore, oltre che per ragioni economiche, anche per l’assenza di tossicità; il g. naturale distribuito per usi domestici viene odorizzato per motivi di sicurezza. Il g. naturale riveste grande importanza anche per l’impiego petrolchimico, basato soprattutto sulla produzione di g. di sintesi e di acetilene che costituiscono, a loro volta, materie prime di base per l’ottenimento di prodotti legati all’industria dei detersivi, dei fertilizzanti, delle materie plastiche, della gomma ecc.; dal g. naturale si ottengono anche nerofumo, cloruro di metile, cloroformio e altri prodotti di notevole interesse commerciale.
Produzione
La maggior parte delle riserve mondiali originarie di g. naturale, oltre il 90%, sono contenute in poco più di 400 grandi giacimenti, mentre la rimanente parte (circa il 6%) è intrappolata in più di 24.000 piccoli giacimenti. Durante l’ultimo ventennio del 20° sec. il maggior paese produttore di g. naturale è stata l’Unione Sovietica e, dopo la dissoluzione di questa, la CSI (Comunità degli Stati Indipendenti). Attualmente il primato spetta alla Russia, con oltre il 20% della produzione mondiale; la regione del Nord America nel suo complesso fornisce più del 25%. In Europa, i principali produttori sono Regno Unito e Norvegia. Dai primi anni Duemila anche paesi dell’area asiatica (Indonesia, Malesia, e, in misura minore, Cina e India) hanno registrato incrementi significativi della produzione di g. naturale. Le riserve di g. naturale nel mondo sono andate storicamente aumentando, a causa di nuove scoperte, principalmente in Iran. Le riserve accertate ammontano, in tutto il mondo, a oltre 120.000 miliardi di metri cubi, ma alcune valutazioni, comprendenti anche le riserve potenziali o ancora in fase di accertamento, si spingono fino a 140-150.000 miliardi di metri cubi. La produzione di g. naturale è passata da 2.227 t (1996) a 2.425 milioni di t (2000), raggiungendo i 3 milioni di t nel 2007.
Nell’Europa occidentale, dove il g. rimane il combustibile maggiormente usato dopo il petrolio, la domanda di g. naturale è in crescita costante (+3,1% ). In prospettiva, i consumi di g. naturale sono previsti in forte aumento, tanto da portare la quota, rispetto ai consumi energetici totali nel mondo, dal 24% del 2003 al 26% nel 2030, secondo le proiezioni della Energy Information Administration americana.
La produzione italiana di g. naturale è stata caratterizzata negli ultimi anni 1990 e nei primi 2000 da una consistente flessione, un trend in atto già dal 1994, dovuta sia alla rarefazione dell’attività di prospezione e di perforazione di nuovi pozzi sia al progressivo impoverimento dello stock di riserve minerarie di g. (dimezzatesi nell’arco degli ultimi tredici anni). L’Italia si avvia dunque a diventare sempre più un paese fortemente dipendente dalle importazioni di g. naturale (nel 2004 l’85% del fabbisogno). Le regioni produttive sono Basilicata, Puglia, Sicilia, Emilia Romagna, Marche, Molise e Abruzzo, mentre in mare la maggiore produzione di g. proviene dall’Adriatico, che fornisce il 53,2% dell’intera produzione nazionale. La ripartizione dei volumi di importazione in relazione alla provenienza mette in evidenza che la maggiore quota di g. naturale importato proviene dalla Russia attraverso i varchi di Tarvisio (gasdotto TAG103) e Gorizia, cui seguono le importazioni dall’Algeria al terminale di Mazara del Vallo, attraverso i gasdotti TTPC104 (via Tunisia) e TMPC (in acque territoriali italiane).
Gas nobili
Si chiamano g. nobili (o rari o inerti) gli elementi elio, neo, argo, cripto, xeno e radon costituenti il gruppo zero (o 18, secondo le più recenti convenzioni) del sistema periodico degli elementi (è possibile preparare, in condizioni particolari, composti di alcuni di questi elementi poco reattivi). Più genericamente si chiama inerte un g. che presenta difficoltà a reagire chimicamente (oltre ai g. nobili spesso si considerano come inerti l’azoto, l’anidride carbonica ecc.). I g. nobili sono presenti nell’aria dalla quale sono estratti anche industrialmente, come sottoprodotti durante il processo di distillazione dell’aria liquida o di preparazione dell’ammoniaca a partire dall’azoto atmosferico; essi trovano impiego nel riempimento di lampade a incandescenza e a luminescenza, di dispositivi elettronici come g. inerti, in alcune lavorazioni chimiche e meccaniche.
Gas di sintesi
Miscela gassosa costituita essenzialmente da idrogeno e da ossido di carbonio, così denominata perché trova largo impiego per la sintesi del metanolo e degli alcoli superiori, per la sintesi Fischer-Tropsch delle benzine, per le reazioni di ossosintesi; dal g. di sintesi, convertendo l’ossido di carbonio in anidride carbonica, che viene poi eliminata per lavaggio con adatte soluzioni assorbenti (carbonati alcalini, etanolammine ecc.), si può ottenere idrogeno puro che è utilizzabile per la sintesi dell’ammoniaca e per l’idrogenazione dei grassi e, in misura minore, dei carboni e degli oli minerali. Il g. di sintesi si ottiene principalmente partendo da combustibili liquidi (benzine a basso numero di ottano, oli combustibili) o gassosi (g. naturali, g. di raffineria, miscele di propano e butano) e sfruttando le reazioni che avvengono fra gli idrocarburi costituenti i combustibili e il vapor acqueo, dalle quali si ottiene, nel campo di temperature generalmente adottate per questi processi, un g. costituito essenzialmente di idrogeno e ossido di carbonio, e contenente quantità, tanto meno rilevanti quanto maggiore è la temperatura, di anidride carbonica e di metano non convertito. Il processo è, nel suo complesso, notevolmente endotermico e i vari procedimenti adottati per la produzione industriale di g. di sintesi si possono classificare in base al modo in cui viene fornito il fabbisogno termico per mantenere la temperatura ai livelli necessari: a) processi ciclici, generalmente catalitici, in cui la reazione si fa avvenire su un catalizzatore (a base di nichel) attraversato alternativamente dalla miscela reagente (idrocarburi e vapor acqueo) e da prodotti caldi di combustione che ripristinano il livello termico della massa catalitica; b) processi continui catalitici, di gran lunga i più adottati, in cui la miscela reagente percorre tubi riempiti di catalizzatore (a base di nichel) e lambiti esternamente da prodotti di combustione che trasmettono alla miscela che reagisce sulla massa catalitica interna il calore necessario a mantenere il livello termico delle reazioni (700-900 °C); i tubi sono sospesi verticalmente in un forno in cui sono installati bruciatori; il processo avviene generalmente sotto pressione (anche fino a 40 bar) e non può trattare miscele di idrocarburi con punti di ebollizione superiori a 220 °C perché altrimenti si avrebbe deposito di carbonio con conseguente disattivazione del catalizzatore; c) processi continui non catalitici, in cui il calore necessario per le reazioni di conversione ossidante è fornito dalla combustione di una parte degli idrocarburi alimentati nel reattore; il comburente è ossigeno puro (e non aria, per evitare la presenza di azoto nel g. risultante); la temperatura è compresa tra 1100 e 1500 °C e il processo, detto anche di combustione (ossidazione) parziale, avviene sotto pressione. Rispetto ai processi continui catalitici il processo di combustione parziale mostra alcuni vantaggi (la maggior flessibilità rispetto alle materie prime impiegate, la capacità di convertire combustibili con elevato contenuto in zolfo senza dover procedere a una desolforazione preliminare, la possibilità di operare a pressioni anche notevolmente superiori ai 40 bar che rappresentano il valore massimo ammissibile dentro ai tubi dei forni catalitici continui), ma presenta l’inconveniente di richiedere un elevato consumo di ossigeno puro con le conseguenti implicazioni sui costi.
La produzione di g. di sintesi tramite gassificazione di combustibili solidi (carboni, coke) rappresenta una percentuale molto modesta della produzione totale e ciò è da mettere in relazione anche agli elevati costi d’impianto; tale produzione, però, potrebbe subire un sostanziale incremento in relazione a una più difficile disponibilità di combustibili di origine petrolifera.
Depurazione dei gas
Nei g. è spesso reperibile la presenza di impurezze sotto forma di particelle (liquide o solide) in sospensione o di componenti gassosi. La depurazione, cioè l’eliminazione delle impurezze, può essere eseguita per vari scopi: per diminuire gli inquinamenti atmosferici togliendo ai g., prima dello scarico nell’atmosfera, quei componenti che potrebbero nuocere o arrecare disturbi agli abitanti dei centri urbani; per evitare, in molti processi chimici, l’avvelenamento dei catalizzatori adottati; per prevenire corrosioni o fenomeni di erosione meccanica ecc. Per quanto concerne i sistemi adottati per eliminare le particelle solide ➔ depolverizzazione; del tutto analoghi i metodi di depurazione per le particelle liquide. La separazione di impurezze gassose viene generalmente eseguita per assorbimento o per adsorbimento o per conversione chimica. Nel primo caso il g. viene lavato con liquidi dotati di proprietà assorbenti selettive verso il componente o i componenti da eliminare (così, per es., l’idrogeno solforato può essere trattenuto da soluzioni di soda caustica, di carbonato sodico, di tioarseniato sodico, di etanolammina, di fenato sodico ecc.). Si ricorre frequentemente all’adsorbimento per diminuire o eliminare l’umidità dei g. (così, per es., i g. naturali, prima del degasolinaggio, sono essiccati con allumina attivata o altro materiale adsorbente). Numerose applicazioni trova anche la depurazione per conversione chimica delle impurezze; così, per es., l’idrogeno solforato contenuto nel g. di distillazione viene eliminato nelle cosiddette casse di depurazione; le ultime tracce di ossido di carbonio contenute nel g. di sintesi dell’ammoniaca possono essere eliminate per metanazione, facendo reagire cataliticamente l’ossido con idrogeno e ottenendo metano che è meno nocivo nei confronti del catalizzatore di sintesi.
Astronomia
G. interstellare
G. (principalmente idrogeno) presente nello spazio tra le stelle in varie forme (ionizzato, atomico, molecolare), con densità molto bassa (circa 0,4 atomi/cm3 nella nostra galassia). La sua distribuzione è estremamente disomogenea. Si conoscono tre componenti principali: idrogeno neutro diffuso (regioni HI); nubi molecolari; nebulose brillanti di idrogeno ionizzato (regioni HII).
Nelle regioni HI l’idrogeno neutro e altri g. interstellari sono distribuiti su tutto il volume del disco galattico, in uno strato alto circa 400 pc, e sono stati studiati inizialmente attraverso le righe di assorbimento che producono negli spettri continui di un gran numero di stelle. Esperimenti spaziali con osservatori ultravioletti hanno permesso di rivelare in questo modo un grande numero di atomi e molecole interstellari (H, C, N, O, Mg, Si, P, S, Ar, Mn, Na, K, Ca, Ti, Fe, H2, HD, CH, CN, CO ecc.) con vari stati di ionizzazione. Lo sviluppo della radioastronomia ha consentito lo studio dettagliato della distribuzione del g. nella galassia, anche in regioni nascoste alle osservazioni visibili dall’assorbimento della polvere interstellare. Fondamentale in questo ambito è lo studio della riga a 21 cm di lunghezza d’onda (corrispondenti a una frequenza ν0=1420,40575 MHz) prodotta dall’idrogeno atomico neutro. La riga osservata da Terra in una certa direzione proviene da molte nubi con differenti velocità, e il suo profilo è interamente determinato dai moti macroscopici di tali nubi poiché i moti del g. interstellare producono, per effetto Doppler, importanti spostamenti dalla frequenza naturale ν0. Attraverso lo studio dei profili di questa riga è stato possibile quindi ricavare non solo la distribuzione del g. ma anche la dinamica dei moti a grande scala della nostra galassia e di alcune galassie esterne. Risulta che l’idrogeno interstellare ruota nel piano galattico su orbite circolari intorno al centro della nostra galassia, con velocità comprese tra 220 e 280 km/s. La densità media nell’idrogeno atomico varia da 0,32 atomi/cm3 a 0,44 atomi/cm3, per distanze dal centro galattico comprese tra 4 e 12 kpc; la densità decresce sia a distanze minori che maggiori. Nella regione centrale della galassia, l’HI è distribuito in un disco inclinato di un angolo di circa 25° rispetto al piano galattico. La massa totale di idrogeno neutro nel disco galattico è di 2,5∙109M⊙. Solo poche molecole semplici sono state rivelate nelle regioni HI: per es., il CO, che è particolarmente stabile e quindi meno facilmente distrutto dalla radiazione UV stellare e dai raggi X cosmici, OH, H2O.
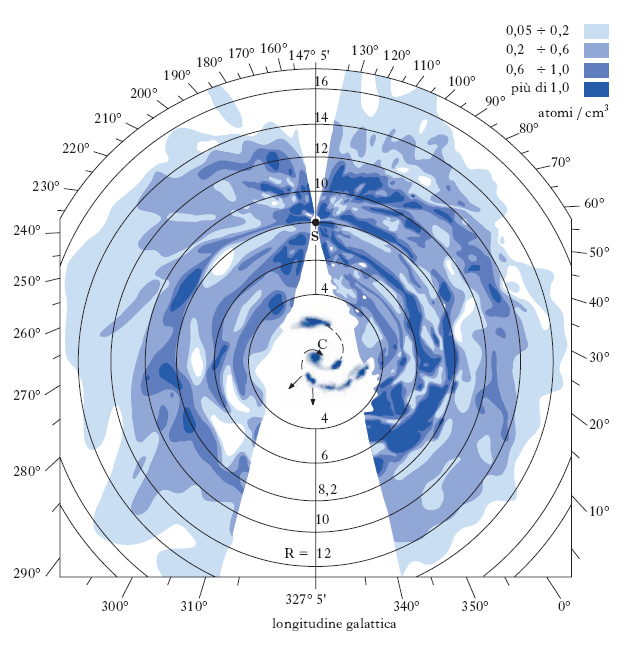
Le nubi molecolari sono concentrazioni molto più marcate di materia interstellare, in cui il g. coesiste con polvere densa che scherma efficacemente la radiazione ultravioletta delle stelle. Così protette dalla fotodissociazione, si possono formare molecole, dalle più semplici alle più complesse. Le nubi molecolari sono concentrate in un anello tra 5 e 8 kpc dal centro (con uno spessore di circa 130 pc) e nella regione centrale della galassia, a distanze inferiori a 1 kpc dal centro. La massa totale di idrogeno molecolare nel disco galattico è di circa 2∙109M⊙: la maggior parte di questo è distribuita in circa 4000 nubi molecolari giganti. Le nubi molecolari più vicine al Sole (fig. 3) sono state studiate in dettaglio ed è stato possibile ricavarne distanza, velocità, massa (tra 104 e 106 M⊙), dimensioni e temperature.
All’interno delle nubi molecolari si formano le stelle; le stelle più giovani hanno una forte emissione UV che ionizza efficacemente l’idrogeno contenuto in una regione abbastanza estesa intorno alla stella (sfera di Strömgren). Si formano così le regioni HII. Queste sono rivelabili sia nel visibile sia nel continuo radio. Importante è inoltre l’emissione infrarossa dai grani di polvere interstellare riscaldati a T∿100 K dall’emissione ultravioletta delle stelle nella regione HII. Le regioni HII sono distribuite sui bracci a spirale, come risulta evidente sia dallo studio di galassie vicine sia dalle osservazioni della nostra galassia, utilizzando osservazioni radio per stimare le loro distanze.
Geologia
G. vulcanici
Costituiscono i prodotti fondamentali dell’attività vulcanica, e si manifestano sia attraverso emissioni violente di vapori acidi rilasciati da vulcani altamente attivi sia nelle emanazioni quasi invisibili di diossido di carbonio dal sottosuolo. Sono prodotti quasi continui dell’attività vulcanica e caratterizzano anche i vulcani in stato di quiescenza. Le eruzioni possono produrre quantità letali di gas tossici, i quali sono responsabili del 3% delle morti causate dall’attività dei vulcani. La composizione dei gas vulcanici dipende dal tipo di vulcano e dal suo stato eruttivo. In ordine di abbondanza, gli elementi più comuni sono H2O, CO2, S2, H2, H2S e CO. Alcuni di essi, quando sono emessi da crateri attivi, reagiscono nell’atmosfera formando aerosol. Possono essere studiati usando molte tecniche, che vanno dal campionamento diretto delle fumarole al remote sensing (telerilevamento). Il monitoraggio delle emissioni gassose dovrebbe essere effettuato con una frequenza sufficiente per delineare l’attività di fondo di ciascun vulcano, rispetto alla quale si possano definire delle soglie di attenzione da utilizzare per la gestione del rischio.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


