Genetica. Screening genetico
Enciclopedia della Scienza e della Tecnica (2007)
Genetica. Screening genetico
Lo screening genetico consiste nella ricerca di individui la cui costituzione genetica (genotipo) è causa determinante o predisponente di malattie nell'individuo stesso oggetto dello screening o nei suoi discendenti. Gli screening genetici vengono suddivisi a seconda della popolazione bersaglio in prenatali, neonatali e dell'età adulta e possono riguardare la totalità di una popolazione o una subpopolazione. Lo screening genetico si distingue nettamente da altri tipi di screening medico poiché comporta implicazioni per i familiari dell'individuo oggetto dello screening, che potrebbero non avere interesse a conoscere la loro costituzione genetica.
Lo screening genetico è un procedimento estremamente delicato poiché accanto a benefici può determinare effetti negativi. I benefici dello screening genetico sono costituiti dalla diagnosi presintomatica di una malattia o di una predisposizione a una malattia, l'evidenziazione di una predisposizione nei confronti di effetti negativi provocati da fattori ambientali e l'identificazione dello stato di portatore di una malattia con implicazioni cruciali per le eventuali scelte riproduttive. Gli effetti negativi possibili sono di natura psicologica (ansietà, perdita di stima di sé, senso di colpa) e sono strettamente correlati alle informazioni ricevute, soprattutto a causa delle difficoltà di comprensione e di interpretazione delle stesse.
In tale contesto, un uso improprio delle informazioni relative a ciò che è stato messo in evidenza attraverso questa metodica può essere all'origine di forme di emarginazione sociale, di discriminazione in ambito lavorativo e può da ultimo determinare rilevanti danni economici.
sommario
1. Principî generali. 2. Screening di neonati. 3. Screening di popolazioni per individuare la suscettibilità genetica a malattie a sviluppo nell'età adulta . 4. Screening di eterozigoti. 5. Screening fetali. □ Bibliografia.
Principî generali
Lo screening prenatale consiste nella identificazione di malattie genetiche nel periodo prenatale attraverso diverse metodologie fra cui studio ecografico del feto, analisi del siero materno per fattori di rischio, esame di cellule fetali ottenute con amniocentesi o con i villi coriali, analisi di cellule fetali nel sangue materno e analisi dell'embrione preimpianto. In particolare gli screening prenatali sono attualmente diretti: (a) alla identificazione con ecografo di numerose malformazioni congenite; (b) alla individuazione di donne a rischio per la sindrome di Down e per altre aberrazioni cromosomiche, tramite valutazione ecografica della translucenza nucale e misure di diversi parametri biochimici nel sangue materno (tra cui β-hCG e PAPP-A nel primo trimestre e α-fetoproteina, hCG totale, estriolo non coniugato e inibina nel secondo trimestre); (c) a definire il rischio di difetto del tubo neurale tramite ecografia e misura della α-fetoproteina.

Lo screening neonatale ha lo scopo specifico di identificare malattie genetiche, specie errori metabolici ed endocrini, per le quali trattamenti opportuni consentono di prevenire lo sviluppo clinico della malattia. Le malattie neonatali attualmente oggetto di screening sono per consenso unanime la fenilchetonuria, l'ipotiroidismo congenito, la sordità congenita e l'anemia falciforme. Oggetto di dibattito è invece lo screening di altri disordini metabolici, fibrosi cistica, iperplasia surrenale congenita, insufficienza mentale legata all'X-fragile e il difetto di G6PD (tab. 1).
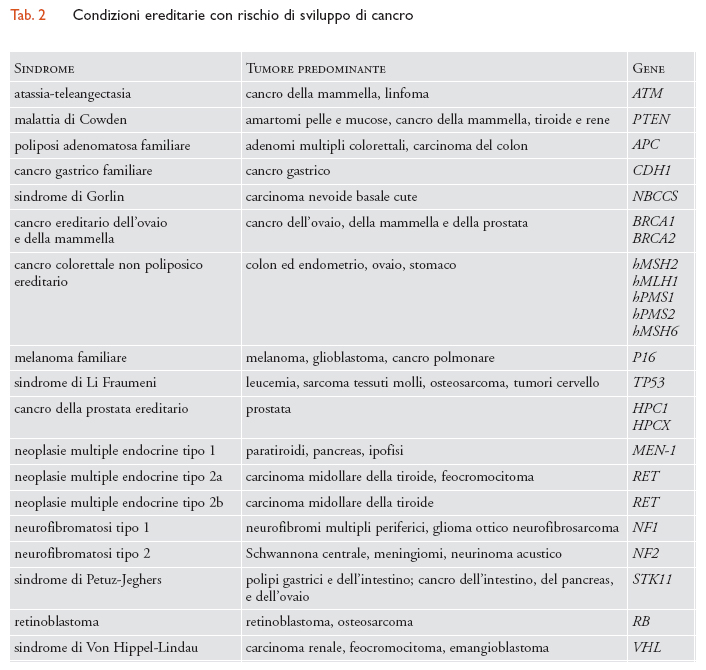
Lo screening genetico in età adulta comprende: (a) individuazione dello stato di portatore di una malattia autosomica recessiva o legata al cromosoma X per prevenire lo sviluppo della malattia nei discendenti, attraverso screening dell'intera popolazione o tramite lo screening a cascata dopo l'identificazione di un caso indice; le malattie attualmente oggetto di screening in popolazioni a rischio sono la malattia di Tay-Sachs, la β-talassemia, l'anemia falciforme, l'α-talassemia e la fibrosi cistica; (b) identificazione di malattie dominanti a esordio nell'adulto o di tumori ereditari in subpopolazioni a rischio; le malattie a insorgenza nell'adulto oggetto di discussione sull'opportunità di includerle in programmi di screening sono ipercolesterolemia familiare, distrofia miotonica, corea di Huntington, rene policistico, emocromatosi ereditaria, trombofilia da eterozigosità per fattore Leyden e difetto di α-antitripsina. Le condizioni ereditarie più comuni con rischio di sviluppo di cancro considerate in programmi di screening sono: atassia-teleangectasia, malattia di Cowden, poliposi adenomatosa familiare, cancro gastrico familiare, sindrome di Gorlin, cancro ereditario dell'ovaio e della mammella, cancro colorettale non poliposico ereditario, melanoma familiare, sindrome di Li Fraumeni, cancro della prostata ereditario, neoplasie multiple endocrine tipo 1, tipo 2a e tipo 2b, neurofibromatosi tipo 1, neurofibromatosi tipo 2, retinoblastoma, sindromi di Peutz-Jeghers e di Von Hippel-Lindau (tab. 2); (c) definizione della predisposizione allo sviluppo di malattie polifattoriali (coronaropatie, diabete tipo I e II, asma allergico, ipertensione arteriosa, malattie cardiovascolari, malattie infiammatorie croniche intestinali, artrite reumatoide, lupus eritematoso sistemico e diverse malattie psichiatriche) e/o di effetti collaterali negativi per la somministrazione di farmaci specifici.
Lo screening genetico è un mezzo appropriato di trattamento medico a condizione che vengano rispettate le seguenti raccomandazioni: razionalità, beneficio superiore ai danni potenziali, accettazione da parte della popolazione bersaglio, fattibilità, prevalenza dei benefici sui costi, educazione della popolazione, laboratori attrezzati, pregressi programmi pilota, serietà della malattia, inserimento nel sistema sanitario, messa a punto di adeguate capacità di seguire longitudinalmente i soggetti individuati, specificità e sensibilità dei test prescelti, partecipazione al programma volontario preceduto da consultazione genetica e consenso informato (può essere non necessario se l'analisi è utile per il soggetto oggetto dello screening), riservatezza assoluta. L'uso dei campioni di sangue per altri scopi necessita per lo più di un consenso informato addizionale specifico.
I progressi della cosiddetta medicina genomica nei Paesi sviluppati, stanno determinando una notevole espansione della medicina preventiva basata su screening di popolazione per determinare la suscettibilità individuale per le cosiddette malattie comuni tra cui le cardiopatie, il diabete, il cancro e le malattie neurodegenerative progressive. L'individuazione delle persone a rischio comporta l'applicazione della prevenzione primaria, tramite, per esempio, provvedimenti dietetici o esercizi fisici specifici, e/o di quella secondaria via intervento farmacologico. Queste applicazioni richiedono nuove raccomandazioni e valutazioni, in specie per quanto riguarda i rapporti costi/benefici. Ovviamente per queste future estensioni sarà ancora più rilevante l'uso del consenso informato ottenuto dopo estensiva consultazione genetica. Raccomandazioni specifiche riguardano lo screening dei bambini e degli adolescenti che, in accordo con le raccomandazioni dell'Accademia Americana di Pediatria (1992), può essere eseguito solo nelle seguenti condizioni: immediato beneficio clinico per il bambino o l'adolescente legato alla possibilità di instaurare misure di prevenzione della malattia o delle sue conseguenze e di ottenere un ritardo dell'esordio o di limitarne la gravità; benefici per altri membri della famiglia, a condizione che non vi siano effetti negativi sul minore.
Screening di neonati
Aspetti generali
Lo screening genetico dei neonati riguarda essenzialmente alcuni errori congeniti del metabolismo tra cui la fenilchetonuria e l'ipotiroidismo congenito (insufficiente produzione di ormone tiroideo). Gli errori congeniti del metabolismo sono un gruppo di malattie genetiche dovute alla deficienza di un enzima specifico che provoca malattia per accumulo di metaboliti a monte della reazione enzimatica coinvolta, o per mancanza del prodotto della reazione. La base razionale per questo screening è costituita dal fatto che numerosi errori del ricambio e l'ipotiroidismo congenito non si manifestano clinicamente nel neonato ma si esprimono solo nel lattante, nel bambino o addirittura nell'adulto quando si sono già verificati danni irreparabili. L'individuazione di queste affezioni nel neonato consente di instaurare un trattamento appropriato e quindi di prevenire o quanto meno attenuare le manifestazioni della malattia. La fenilchetonuria (PKU), una malattia dovuta al difetto del metabolismo di un amminoacido, la fenilalanina, fu la prima malattia oggetto di screening neonatale che iniziò in molte popolazioni fin dagli anni Sessanta del Novecento. Attorno agli anni Settanta allo screening per la PKU venne associato lo screening per l'ipotiroidismo congenito.
Le linee guida per lo screening neonatale variano nei diversi Paesi sviluppati. In Europa il programma minimo riguarda la PKU e l'ipotiroidismo. In gran parte della Germania e della Svizzera tuttavia sono oggetto di screening anche altri rari errori metabolici. Negli USA i programmi variano da uno Stato all'altro. Di recente l'American Academy of Medical Genetics ha raccomandato l'applicazione nello screening del neonato di una nuova tecnologia, la gas cromatografia-spettrometria di massa, che consente di identificare più di venti disordini metabolici relativamente rari, a eccezione del difetto della deidrogenasi degli acidi grassi a media catena, appartenenti al gruppo dei difetti del ricambio degli acidi grassi e delle acidurie organiche. Per queste altre malattie metaboliche vi sono notevoli riserve legate alla rarità, alla marcata eterogeneità sia clinica che genetica, alla mancanza di dati sul vantaggio della diagnosi precoce e all'assenza di un rapporto positivo costi/benefici. L'opportunità di screening per l'anemia falciforme, il difetto di α-1- antitripsina, l'iperplasia surrenale congenita (praticato in numerosi Stati americani, in Svezia e Olanda), le distrofinopatie (praticato solo in Inghilterra) e la fibrosi cistica è ancora oggetto di dibattito. Nei Paesi in via di sviluppo lo screening neonatale genetico non è operante oppure sono in corso limitati programmi pilota. Questa situazione è in contrasto con uno dei principî etici più importanti per regolare gli screening genetici costituiti dalle pari opportunità di accesso da parte di tutte le popolazioni. Nei prossimi paragrafi affronteremo in dettaglio lo screening della PKU, dell'ipotiroidismo e della fibrosi cistica e faremo un cenno allo screening per la sordità congenita, l'anemia falciforme, il difetto in G6PD e l'insufficienza mentale legata al sito X-fragile. Lo screening per le distrofinopatie si fonda sulla determinazione della creatin-fosfochinasi (CPK) nel sangue seguita in caso di positività dall'analisi mutazionale della distrofina (identificazione preclinica di distrofia muscolare progressiva tipo Duchenne e Becker). Nello screening per l'iperplasia surrenale congenita si dosa il 17-idrossiprogesterone plasmatico che è aumentato nel caso del difetto enzimatico più comune (21 idrossilasi) causa della malattia. In presenza di positività si pratica un'analisi più completa degli steroidi plasmatici seguita da analisi mutazionale.
Malattie principali oggetto di screening
Iperfenilalaninemie compresa la fenilchetonuria. - Le iperfenilalaninemie sono difetti congeniti della idrossilazione della fenilalanina a tirosina. L'incidenza media è di 1 caso su 12.000 nati vivi. Le iperfenilalaninemie hanno un genotipo estremamente eterogeneo e sono dovute a mutazioni di diversi geni codificanti proteine componenti della reazione idrossilasica. La forma più comune è la iperfenilalaninemia da difetto di fenilalanina-idrossilasi (fenilchetonuria, PKU). La PKU è molto eterogenea geneticamente; si conoscono infatti più di 400 mutazioni diverse, che mostrano una distribuzione specifica per ciascuna popolazione. Il test di screening neonatale misura la fenilalanina con un saggio semiquantitativo basato sulla inibizione della crescita batterica determinata da livelli elevati di fenilalanina. Metodi alternativi sono rappresentati dalla determinazione fluorimetrica, enzimatica o tramite la gas cromatografia-spettrometria di massa. La PKU non trattata provoca un quadro di grave ritardo mentale associato talvolta ad autismo (difficoltà ai rapporti interpersonali), iperattività, convulsioni, comportamento aggressivo, eczema e, di frequente, aspetto chiaro della pelle e dei capelli.
In presenza di livelli elevati di fenilalanina si instaura una dieta povera di fenilalanina. La dietoterapia deve essere seguita attentamente e rivalutata criticamente alla luce dei controlli periodici di fenilalanina nel sangue. Per avere buoni risultati la dieta deve essere instaurata prima delle tre settimane di vita. Il trattamento previene l'insufficienza mentale e le alterazioni neurologiche. Vi è oramai accordo sul concetto secondo cui la dieta deve essere continuata per tutta la vita. Un vantaggio addizionale di questo atteggiamento è la prevenzione dei danni (microcefalia, difetto congenito di cuore, ritardo di crescita intrauterina) provocati nel feto non PKU di madre PKU non sottoposta a dieta con bassa fenilalanina. Queste alterazioni sono dovute all'effetto negativo sugli organi fetali della fenilalanina in eccesso proveniente dalla madre. In ogni caso di PKU individuato con le metodologie tradizionali occorre completare la diagnosi con la ricerca della mutazione specifica. Questa informazione ha utilità pratica per l'individuazione nell'ambito della famiglia dei portatori e per la diagnosi prenatale.
Ipotiroidismo congenito. - L'ipotiroidismo congenito ha un'incidenza di 1:4000 nati vivi. Le cause sono difetti dell'embriogenesi risultanti in agenesia o disgenesia della tiroide (si tratta per lo più di forme sporadiche non ereditarie); difetti metabolici congeniti della biosintesi degli ormoni tiroidei; difetti congeniti o errori metabolici dell'ipofisi o dell'ipotalamo con conseguente ridotta produzione dell'ormone tireotropo. Nella maggior parte dei casi l'eziologia è sconosciuta. Poiché per lo sviluppo del sistema nervoso centrale è necessario l'ormone tiroideo, ogni ritardo diagnostico anche breve nella diagnosi dell'ipotiroidismo determina un danno permanente del SNC con conseguente insufficienza mentale. Su queste basi razionali, programmi di screening per l'ipotiroidismo congenito sono operanti da diversi anni in Europa, Giappone e Stati Uniti d'America. Il materiale da esaminare è costituito da campioni di sangue raccolti su carta da filtro al quinto giorno di vita. Due diverse strategie sono attualmente in uso: negli USA viene misurata la tiroxina serica, cioè il principale ormone tiroideo (T4), che dovrebbe consentire di diagnosticare tutti i casi di ipotiroidismo eccetto le forme rarissime dovute a resistenza periferica agli ormoni tiroidei; in Europa viene misurato il TSH (Thyroid stimulating hormone, ormone che stimola la tiroide). Un suo aumento consente di identificare le forme più comuni di ipotiroidismo da difetto della tiroide; con questo approccio sfuggono i casi rari da difetto dell'ipofisi o dell'ipotalamo. L'esame del TSH è però molto più sensibile della misurazione di T4. Con entrambi i metodi si hanno falsi positivi e negativi. Ovviamente i migliori risultati sono ottenibili dalla valutazione contemporanea di T4 e TSH. I casi positivi devono essere confermati da una valutazione completa della funzione tiroidea. Dopo conferma diagnostica, il trattamento con T4 deve essere avviato immediatamente. Quando la terapia viene iniziata entro il primo mese di vita i risultati sono eccellenti.
Sordità congenita. - Il difetto uditivo è una delle più comuni anomalie congenite svelabili alla nascita, riguardante da 1-3 neonati per 1000 nati vivi. La diagnosi avviene abitualmente a 14 mesi circa con conseguenze negative sullo sviluppo del linguaggio e di quello cognitivo. Di recente nei programmi di screening neonatali è stato incluso lo screening dei difetti uditivi tramite lo studio delle emissioni otoacustiche evocate (EOAE) o con l'analisi dei potenziali uditivi evocati (ABR) sia singolarmente che in combinazione. Entrambe le metodologie sono prive di invasività, rapide (richiedono solo 5 minuti) e di facile esecuzione. L'identificazione precoce seguita da intervento appropriato, come l'impianto cocleare, consente di prevenire queste conseguenze. Occorre eseguire un secondo screening entro il primo mese di vita. Viene inoltre raccomandato che i casi risultati positivi allo screening vengano studiati con analisi del DNA per mutazioni del gene che codifica per la connessina 26, presente nell'orecchio interno, il cui difetto è causa di circa il 50% dei casi di sordità ereditaria non sindromica.
Anemia falciforme. - L'anemia falciforme è una delle più frequenti malattie genetiche con una incidenza particolarmente elevata in Africa equatoriale, nella Penisola Arabica e in India. Ovviamente, specie per la tratta degli schiavi, avvenuta in passato, si ha una elevata incidenza anche in America del Nord e del Sud e nei Caraibi. In Italia l'anemia falciforme si osserva in Sicilia e Calabria. Occorre tuttavia tenere presente che lo stato di portatore per l'HbS è comune in molte popolazioni africane da cui provengono immigrati nel nostro Paese. Il genotipo più comune alla base dell'anemia falciforme è l'omozigosi per l'allele HbS, risultante da una singola sostituzione nucleotidica che determina la sostituzione dell'acido glutammico con la valina nel codone 6 del gene che codifica per la β-globina, componente essenziale della molecola emoglobinica dell'adulto. Meno di frequente l'anemia falciforme è dovuta alla presenza di combinazioni genetiche (eterozigoti composti) tra l'HbSe altri difetti quantitativi e qualitativi delle β-globine(β0 o β+-talassemia, HbC e altre rare emoglobinopatie).
La falcemia provoca anemia emolitica e crisi di occlusione vascolare. L'anemia falciforme comporta, inoltre, fin dall'età del lattante diverse complicazioni potenzialmente fatali, tra cui sepsi iperacuta specie da Streptococcus pneumoniae e crisi di sequestrazione splenica. L'identificazione neonatale consente di mettere in opera un programma di sorveglianza e di cura, inclusa la profilassi penicillinica, le vaccinazioni contro pneumococco, meningococco e Haemophilus influentiae e il trattamento energico delle crisi occlusive e delle infezioni che appare ridurre la morbilità dei bambini affetti e consentire una migliore sopravvivenza. Queste considerazioni rappresentano la base razionale per lo screening neonatale. Tuttavia non è ancora stabilito con esattezza se lo screening per l'anemia falciforme abbia in effetti un impatto positivo su morbilità e mortalità. Lo screening viene effettuato tramite elettroforesi o isoelettrofocusing dell'emoglobina.
La conferma dei risultati ottenuti con questi metodi viene fatta con la ricerca delle mutazioni specifiche sul DNA (HbS e mutazioni β-talassemiche) o con la cromatografia ad alta pressione in fase liquida. Questi metodi consentono di identificare per lo più anche le β-talalassemie, le α-talassemie e altre emoglobinopatie di rilevanza clinica. L'identificazione di un caso di anemia falciforme comporta la necessità di studiare approfonditamente tutta la famiglia. Lo screening per anemia falciforme è operativo negli USA e a Cuba per tutti i neonati e in Inghilterra e Francia per i gruppi a rischio. Ovviamente tale screening comporta importanti rischi specie quando condotto in popolazioni in cui i negri rappresentano una minoranza etnica. Tra questi rischi occorre menzionare in particolare la stigmatizzazione, la riduzione della stima di sé e la discriminazione.
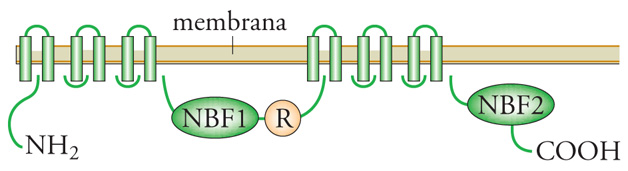
Fibrosi cistica. - La fibrosi cistica (FC) è la malattia più comune a eredità autosomica recessiva nei bianchi caucasici in cui si verifica con una incidenza di 1 caso su 2000-3000 nati vivi. I sintomi principali sono malattia polmonare cronica ostruttiva (da particolare densità del muco), infezioni polmonari persistenti specie da Pseudomonas aeruginosa e Burkolderia cepacia (un germe opportunistico ambientale), ileo da meconio (occlusione intestinale da meconio denso), insufficienza pancreatica e accrescimento difettoso. La diagnosi, sospettata clinicamente, è confermata dal riscontro di valori elevati di cloro nel sudore (>60 meq/l). La fibrosi cistica è dovuta a un difetto molecolare di un canale per il cloro (CFTR, Cystic fibrosis transmembrane conductance regulator) (fig. 2). La fibrosi cistica è geneticamente eterogenea. La mutazione più frequente (circa il 70% in Nord Europa e il 50% in Italia) causa di FC è la delezione di tre paia di basi codificanti fenilalanina al codone 508 (F508del). Oltre la F508del si conoscono oltre 1000 mutazioni circa, alcune delle quali relativamente frequenti e altre osservate solamente in famiglie singole. La distribuzione delle diverse mutazioni è popolazione-specifica. Il trattamento si fonda su drenaggio posturale, fisioterapia, cicli di antibiotici per limitare i danni delle infezioni polmonari e terapia sostitutiva enzimatica pancreatica. La prognosi è migliorata notevolmente. La sopravvivenza media è di circa 30 anni.
L'età media alla diagnosi di FC è di 15 mesi circa, a esclusione dei casi manifestatisi con ileo da meconio o di quelli con storia familiare positiva. Nel primo anno di vita i pazienti affetti da FC vanno incontro a marcato difetto di crescita sia in peso che in statura, edema da ipoproteinemia, colonizzazione polmonare da Pseudonomas aeruginosa, malnutrizione. Di recente diversi studi, di cui almeno due randomizzati, hanno consentito di stabilire che l'individuazione neonatale tramite screening, con successiva diagnosi definitiva entro il quinto mese, comporta importanti benefici sotto il profilo nutrizionale e cognitivo. Lo screening consente inoltre l'individuazione della coppia a rischio con cui avviare la consultazione genetica prima dell'inizio della seconda gravidanza. La tecnica in uso consiste nell'analisi della tripsina serica con metodo immunologico (IRT) sul campione di sangue raccolto su carta da filtro. Nei campioni risultati positivi per l'aumento dei valori di tripsina viene eseguita l'analisi mutazionale del gene CFTR per le mutazioni più comuni popolazione-specifiche.
Come per tutti gli screening genetici vi sono dei limitati effetti negativi dovuti alla presenza di falsi positivi e negativi allo screening iniziale fatto con la determinazione dei livelli di tripsina immunoreattiva. Lo screening neonatale per identificare la CF è oggetto di controversia; attualmente è eseguito in Italia, in alcune regioni della Gran Bretagna e della Francia e in alcuni Stati americani. Sulla base dei dati su esposti l'American Academy of Medical Genetics ha raccomandato di recente l'inclusione dello screening per la CF tra i classici screening neonatali.

Insufficienza mentale legata all'X-fragile. - La sindrome dell'X-fragile è la più frequente causa di insufficienza mentale ereditaria (dopo la sindrome di Down) ed è dovuta all'espansione in generazioni successive di una tripletta polimorfica costituita dai nucleotidi CGG localizzata nel gene FMR-1 (Fragile mental retardation-1) che mappa nel cromosoma X. La proteina codificata da FMR-1 controlla l'attività di numerosi RNA-messaggeri condizionando la plasticità delle sinapsi (base cellulare dell'apprendimento e della memoria a lungo termine). L'incidenza è di 1 caso su 4000 maschi e di 1 su 7000 femmine. Si distinguono due tipi di mutazione: premutazione e mutazione completa. La premutazione consiste in una modesta espansione della tripletta (replicata da 55 a 200 volte) e non è associata a ritardo mentale. La mutazione completa (più di 200 volte) è caratterizzata da una maggiore espansione della tripletta ed è associata a ritardo mentale, modeste note dismorfiche e criptorchidismo (fig. 3). Tra questi due estremi vi è una zona (tra 45 e 60 ripetizioni) di incertezza diagnostica. Studi recenti hanno dimostrato che anche i portatori di premutazione possono avere dei problemi clinici: nelle donne modeste note dismorfiche tipiche della sindrome, turbe della personalità e insufficienza ovarica prematura; nei maschi una sindrome neurologica caratterizzata da tremori, atassia e demenza a insorgenza dopo i 50-60 anni. La premutazione si può espandere a mutazione completa solo per via materna. La diagnosi pre- e postnatale e l'identificazione dei portatori della premutazione si fondano sull'analisi diretta del DNA. Lo screening per la sindrome dell'X-fragile può essere eseguito con diversi metodi che si basano sull'amplificazione del DNA. Lo screening neonatale non ha alcuna giustificazione poiché non è di alcun vantaggio per il neonato; la sua utilità consiste unicamente nella precocità della diagnosi e quindi della consultazione genetica della famiglia. Nella maggior parte dei Paesi occidentali lo screening è attualmente limitato a individui con sintomi suggestivi per la sindrome dell'X-fragile, a coppie appartenenti a famiglie con casi accertati della sindrome e a persone con una storia familiare di difetto mentale che richiedono una consultazione genetica.
Difetto di glucosio-6-fosfato-deidrogenasi (G6PD). - La glucosio-6-fosfato-deidrogenasi è un enzima ubiquitario, che catalizza la prima tappa dello shunt dell'esoso-monofosfato, via principale per la produzione di NADPH. Il difetto di G6PD è la più comune enzimopatia ereditaria. Le incidenze più elevate si hanno in Africa tropicale, subtropicale e in alcune zone del Mediterraneo. Le manifestazioni cliniche più comuni sono l'ittero neonatale e l'anemia emolitica acuta. Questa è scatenata dalla ingestione di fave, dall'assunzione di un gruppo di farmaci ossidanti e da infezioni tra cui quella tifoidea e l'epatite acuta. Si conoscono più di 400 mutazioni del gene che codifica per l'enzima G6PD che producono varianti la cui funzionalità presenta differenti livelli di compromissione. Alcune di queste sono associate ad anemia emolitica cronica.
Lo screening per il difetto in G6PD nel neonato è ancora oggetto di controversia, poiché per esso non sono presenti molti dei requisiti citati in precedenza. Tuttavia in alcuni Paesi a elevata incidenza, quali la Sardegna, la Malesia e la Thailandia è in atto da molti anni. L'utilità dello screening è limitata all'informazione sul rischio di esporsi ad agenti scatenanti l'emolisi, mentre non è ovviamente possibile la prevenzione dell'ittero neonatale. Grazie anche allo screening, in questi Paesi l'incidenza dell'emolisi acuta da difetto in G6PD si è ridotta drasticamente. Lo screening, da effettuare con mezzi poco costosi, deve essere limitato agli omozigoti ed emizigoti, in quanto gli eterozigoti non possono essere evidenziati con metodi semplici. Attualmente si potrebbe utilizzare l'analisi mutazionale per le mutazioni popolazione-specifiche che consentirebbe anche l'identificazione degli eterozigoti.
Screening di popolazioni per individuare la suscettibilità genetica a malattie a sviluppo nell'età adulta
Aspetti generali
In questi ultimi anni, grazie ai notevoli progressi compiuti dalla genetica molecolare si è sviluppato un dibattito sull'opportunità di eseguire uno screening di popolazione per malattie a insorgenza nell'adulto tra cui l'ipercolesterolemia familiare, la distrofia miotonica, la corea di Huntington, il rene policistico, l'emocromatosi ereditaria, la trombofilia dovuta allo stato eterozigote per il fattore V Leyden.
Per la gran parte di queste malattie è oggi possibile anche l'identificazione delle mutazioni causali che è, da un lato, utile per affinare la prognosi del malato e, in taluni casi, per intraprendere provvedimenti terapeutici individualizzati, mentre dall'altro offre la possibilità di individuare i familiari a rischio di sviluppare la malattia. Con lo stesso tipo di analisi mutazionale o con analisi di associazione tra la malattia e polimorfismi del DNA ‒ segmenti corti di DNA ripetuti in tandem con un numero variabile di ripetizioni in ciascun individuo (microsatelliti) o variazioni polimorfe di un singolo nucleotide (Single nucleotide polymorphism, SNP) ‒ si possono anche identificare le variazioni nucleotidiche di geni che comportano la predisposizione allo sviluppo delle malattie comuni polifattoriali (dovute a interazione tra diversi geni e fattori ambientali) tra cui le coronaropatie, il diabete tipo I e II, l'asma allergico, l'ipertensione arteriosa, le malattie cerebro-vascolari, le malattie infiammatorie croniche intestinali, l'artrite reumatoide, il lupus eritematoso sistemico e diverse malattie psichiatriche (schizofrenia, sindrome bipolare).
Per alcune di queste malattie, come per esempio l'ipercolesterolemia familiare (fattore proaterosclerotico) e il difetto di α-1-antitripsina (causa di enfisema polmonare) l'individuazione in fase preclinica consente di mettere in atto misure preventive (dieta ed esercizio fisico nell'ipercolesterolemia, astensione dal fumo ed evitare ambienti di lavoro dannosi, quali le miniere di carbone, nel difetto di α-1-antitripsina) che possano ridurre la morbilità e la mortalità precoce associata a queste condizioni, anche se tale eventualità non è stata ancora dimostrata con studi longitudinali. In altre malattie appartenenti a questo gruppo, come la corea di Huntington, invece, l'individuazione preclinica è priva al momento attuale di qualunque utilità. La necessità dello screening relativo è pertanto oggetto di controversia. Gli svantaggi di questo tipo di screening sono rappresentati dagli effetti psicologici negativi per coloro che sono stati individui portatori del gene affetto e le possibili discriminazioni sociali. Tale screening, che, dopo l'individuazione di un soggetto malato, è ovviamente del tipo a cascata nel contesto familiare, deve essere preceduto da una adeguata consultazione genetica, nella quale vengano valutati attentamente i pro e contro del procedimento diagnostico. Numerosi adulti dopo la consultazione, specie per malattie per la cura delle quali non vi è trattamento, preferiranno non sottoporsi all'esame.
Un vantaggio di questo tipo di screening, peraltro limitato a coloro che risultano non avere la mutazione in causa, è quello di raggiungere uno stato di tranquillità relativamente alla propria salute, sotto questo profilo specifico. L'esame di screening per queste condizioni è in genere assolutamente controindicato, nel bambino e nell'adolescente, se non quando sia di immediato beneficio per la persona oggetto dello screening. A parte i possibili effetti negativi, la possibilità di incomprensione del problema e lo scarso interesse, lo screening in età precoce elimina anche la possibilità futura di una scelta autonoma nell'accettare di sottoporsi all'esame. Uniche eccezioni in proposito riguardano adolescenti con gravidanza in corso o già pianificata, oppure quei casi in cui l'analisi del bambino è di immediato beneficio per altri membri della famiglia ma nel contempo non è di danno per il bambino stesso oggetto dello screening. Tra le malattie menzionate solamente per l'emocromatosi ereditaria (HFE-1, la forma più comune dell'adulto) ‒ accumulo di ferro in diversi parenchimi ‒ e la trombofilia dovuta a eterozigosi per il fattore V Leyden (mutazione del fattore V che riduce l'effetto anticoagulante della proteina C), l'opportunità dello screening di popolazioni è presa in considerazione ma è oggetto di intenso dibattito. Per l'emocromatosi attualmente è di uso comune solamente lo screening a cascata, vale a dire l'analisi di tutti i familiari di malati o portatori (tramite l'analisi mutazionale associata allo studio del ricambio del ferro). Dalla maggior parte dei ricercatori non viene attuato lo screening per il fattore V Leyden sia nella popolazione normale che in popolazioni ad alto rischio, come donne che usano contraccettivi o in gravidanza.
Suscettibilità allo sviluppo di tumori
Le ricerche di questi ultimi anni hanno chiaramente dimostrato che lo sviluppo di un tumore è determinato dalla presenza dell'alterata funzione di uno o più geni del gruppo degli oncogeni (favorenti lo sviluppo del tumore) e/o degli oncosoppressori (inibenti lo sviluppo del tumore). Lo screening di popolazione o subpopolazione ad alto rischio per la identificazione di portatori di geni predisponenti al cancro è una eventualità futura che è già oggetto di dibattito. Attualmente la ricerca della predisposizione al cancro viene limitata alle famiglie che mostrano almeno un malato affetto da tumore ereditario. La possibilità di un tumore ereditario viene suggerita dalla presenza delle seguenti condizioni in uno o più membri della famiglia: (a) cancro a insorgenza a età insolitamente giovane per quel tipo di tumore; (b) sviluppo bilaterale o multifocale in un singolo organo; (c) sviluppo di più di un tumore primario di ogni tipo in un individuo; (d) sviluppo di cancro in un individuo o in una famiglia in cui vi siano membri con anomalie congenite; (e) famiglie in cui sia stata già identificata una mutazione causa di cancro ereditario.
L'analisi mutazionale di geni implicati nello sviluppo di tumori può essere eseguita per effettuare la diagnosi di cancro ereditario in un individuo affetto e/o per identificare i familiari a rischio dopo l'identificazione della mutazione in un familiare. Lo studio dei familiari deve essere ovviamente preceduto da un'accurata consultazione genetica che illustri i vantaggi, per esempio applicazione di metodiche preventive, e gli svantaggi (stigmatizzazione, perdita di stima di sé). Ovviamente, è necessario richiedere il consenso informato che illustri diversi punti cruciali, fornendo informazioni sul tipo di test, sulle implicazioni del risultato positivo/negativo, possibilità che il test non sia informativo, sul rischio di trasmissione del difetto e sulla possibilità che vi siano ripercussioni psicosociali. Per quanto riguarda lo studio di bambini e adolescenti valgono le considerazioni sopra esposte per le malattie ereditarie a insorgenza tardiva.
Screening di eterozigoti
Aspetti generali

Un secondo tipo di screening genetico è costituito dallo screening di popolazione per individuare gli eterozigoti per una malattia genetica autosomica recessiva o recessiva legata all'X; i soggetti così identificati vengono avviati alla consultazione genetica. I programmi di identificazione di eterozigoti conducono alla definizione di coppie a rischio di generare un figlio malato, prima della nascita di un probando (soggetto affetto dalla malattia); per tale motivo hanno una marcata efficacia preventiva. I requisiti unanimemente ritenuti necessari per intraprendere uno screening genetico di eterozigoti sono i seguenti: (a) la malattia deve avere una elevata frequenza nella popolazione oggetto dello screening, deve essere grave e non deve esistere nella maggior parte dei casi una cura definitiva; (b) devono essere disponibili dei metodi semplici e di basso costo per l'individuazione degli eterozigoti, con un limitato numero di falsi positivi e possibilmente anche di falsi negativi; (c) devono esistere alternative riproduttive sia per una coppia a rischio di malattie autosomiche recessive che per una coppia in cui la madre sia portatrice di una malattia recessiva legata all'X. Le caratteristiche generali e i requisiti per i programmi di screening degli eterozigoti vengono riassunti nella tab. 3.
La maggior esperienza in questo campo della genetica preventiva riguarda la malattia di Tay-Sachs, la β-talassemia e, più di recente, l'anemia falciforme, l'α-talassemia e la fibrosi cistica.
Malattia di Tay-Sachs
La malattia di Tay-Sachs è una delle gangliosidosi GM2. Si tratta di una malattia neurodegenerativa progressiva che porta per lo più a morte entro i primi 4 anni di vita, particolarmente frequente negli ebrei aschenaziti. I portatori possono essere identificati tramite la determinazione dell'attività enzimatica β-esosamminidasica nel siero. La stessa tecnica applicata alle cellule amniotiche o ai villi coriali consente di fare la diagnosi prenatale. Attualmente sia la diagnosi di portatore che quella prenatale vengono confermate con l'analisi diretta delle mutazioni. Nel 1970 negli USA iniziò un programma di screening e consultazione genetica rivolto a persone in età riproduttiva appartenenti alla popolazione ebraica in cui l'incidenza della malattia di Tay-Sachs è di 1:4000 nati vivi e quella dell'eterozigote di 1:30 persone. Questo tipo di programma è stato applicato a numerose comunità ebraiche nel mondo. Le coppie così identificate vennero avviate a una consultazione genetica di tipo non direttivo, in cui vengono discusse le varie opzioni riproduttive inclusa la diagnosi prenatale. La gran parte delle coppie consultate scelse la diagnosi prenatale e l'interruzione della gravidanza in presenza di feto affetto. L'efficacia della prevenzione è stata pari al 90% circa dei casi.
β-talassemia
La β-talassemia è una delle più comuni malattie autosomiche recessive ed è dovuta a difettosa (β+) o assente (β0) sintesi delle catene β della molecola emoglobinica. La malattia è molto frequente nelle popolazioni del bacino del Mediterraneo e particolarmente in Sardegna e a Cipro, nel Medio Oriente, nel Subcontinente indiano e nell'Estremo Oriente. L'emigrazione e la tratta degli schiavi hanno tuttavia portato la β-talassemia in ogni parte del mondo compresi Europa Settentrionale, USA, Canada, Sud Africa e Australia. L'omozigote ha una grave anemia dovuta prevalentemente a distruzione nel midollo osseo dei precursori dei globuli rossi (eritropoiesi inefficace) e in parte minore a emolisi periferica. Se non curati i pazienti con β-talassemia muoiono nella prima infanzia. Il trattamento dello stato omozigote consiste in trasfusioni continuative e terapia chelante (Desferrioxamina B e Deferiprone) per eliminare il ferro accumulato in eccesso a causa delle trasfusioni. La sopravvivenza con questo trattamento si estende fino ai 30 anni. Valida alternativa è costituita dal trapianto di midollo per lo più da fratello HLA compatibile, che porta a guarigione nell'90-95% circa dei casi.
La β-talassemia è eterogenea geneticamente. Si conoscono al momento attuale più di 200 diverse mutazioni. In ogni popolazione, tuttavia, il numero di mutazioni è limitato. Il portatore di β-talassemia, identificato tramite analisi degli indici eritrocitari (volume globulare e concentrazione emoglobinica media ridotta) e determinazione quantitativa dell'HbA2 (aumentata) con metodo cromatografico o elettroforetico, viene studiato con metodi molecolari per definire il tipo di mutazione. La stessa metodologia molecolare viene utilizzata per la diagnosi prenatale sul DNA dei villi coriali o delle cellule amniotiche.
A metà degli anni Settanta programmi di screening per la β-talassemia, associati a consultazione genetica sono stati introdotti in Sardegna, a Cipro, nella Grecia continentale e in alcune minoranze etniche in Gran Bretagna (Ciprioti, Pakistani). Successivamente lo screening si è esteso all'Italia continentale, alla Sicilia e a minoranze etniche a rischio in altri Paesi europei e negli USA. Programmi limitati a coppie con gravidanza in corso sono stati di recente introdotti anche in Paesi in via di sviluppo (Thailandia, Malesia, Cina meridionale, India). In Sardegna, per esempio, con questi programmi si è verificata una prevenzione del 90-100% dei casi. I casi residui sono risultati in prevalenza dovuti a mancata informazione e, meno di frequente, a continuazione di gravidanza di feti omozigoti per ragioni morali, a errori diagnostici di diagnosi di portatore o prenatale o a falsa paternità.
α-talassemia e anemia falciforme
L'α-talassemia, una condizione estremamente diffusa in Africa, nelle regioni del bacino del Mediterraneo e in specie nel Sud-est asiatico, è per lo più dovuta a delezioni di geni strutturali codificanti per l'α-globina. Programmi di screening diretti all'individuazione dei portatori, limitati per lo più a donne con gravidanza in corso, sono operanti in molte regioni del Sud-Est asiatico con lo scopo di individuare le coppie di portatori per la prevenzione dell'idrope fetale (delezione di tutti e 4 i geni). In alcuni Stati americani lo screening riguarda sia i portatori manifesti che quelli silenti con lo scopo di identificare coppie a rischio sia per idrope fetale che per malattia da HbH (delezione di 3 geni). Per quanto riguarda l'anemia falciforme, si rimanda al paragrafo sullo screening dei neonati per le notizie fondamentali sulla clinica e patologia molecolare della malattia. Programmi di screening di portatori, limitati a donne in gravidanza in corso, sono operanti in diversi Paesi europei e in numerosi Stati americani e in Canada limitatamente alle minoranze etniche.
Fibrosi cistica
Un programma di screening della popolazione in età riproduttiva per la prevenzione della malattia è giustificato dal fatto che la fibrosi cistica è la malattia genetica più frequente nella popolazione caucasica, che presenta nella forma classica un fenotipo grave per il quale non esistono cure definitive. D'altro canto vi sono alcuni ostacoli ad attuare uno screening generale, dovuti essenzialmente al fatto che non esistono metodi poco costosi per diagnosticare il portatore sano e alle conseguenze psicologiche prodotte da diagnosi non conclusive. L'unico test di screening è molecolare e si basa sulla ricerca delle mutazioni del gene CFTR più frequenti nella popolazione in esame. Come già detto in merito agli screening neonatali, le mutazioni del gene CFTR sinora descritte sono più di 1000 e anche se ogni singola popolazione presenta uno spettro mutazionale proprio, l'efficienza diagnostica del test genetico risulta molto spesso troppo bassa per l'avvio di un programma di screening dell'adulto. Negli Stati Uniti il National Institute of Health (NIH), l'American College of Medical Genetics (ACMG) e l'American College of Obstetricians and Gynecologists (ACOG) hanno prodotto delle linee guida dove si raccomanda che lo screening sia offerto agli adulti con familiarità per la fibrosi cistica, allo sposo/sposa di pazienti con fibrosi cistica, alle coppie di origine caucasica che attendono un figlio e a quelle che intendono intraprendere una gravidanza, a spose di pazienti con agenesia bilaterale dei vasi deferenti, mentre sia reso solo disponibile alla popolazione generale. Da numerosi programmi pilota portati avanti negli ultimi anni, infatti, si evince che l'adesione a tali programmi è massima nelle categorie più motivate (coppie in attesa di un figlio e parenti di pazienti affetti), mentre è molto scarsa nella popolazione generale. Inoltre lo screening deve essere disponibile, ma non offerto, anche per le coppie di popolazioni nelle quali la malattia è meno frequente: infatti la frequenza del portatore sano varia da popolazione a popolazione: 1/25 nei Caucasici, 1/60 negli Afroamericani, 1/46 negli Ispanici, 1/90 negli Asiatici, 1/29 negli ebrei aschenaziti.
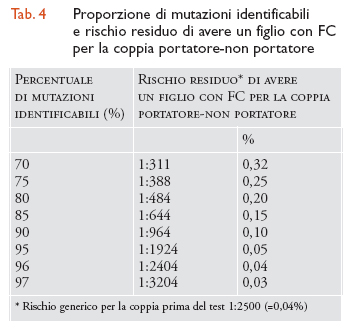
L'efficienza diagnostica del test è limitata essenzialmente dalla impossibilità di trovare tutte le mutazioni del gene CFTR e varia in maniera consistente da popolazione a popolazione. Usando un pannello standard che prevede la ricerca delle 25 mutazioni più frequenti, nelle popolazioni di origine nord-europea l'efficienza è di circa il 90% e negli ebrei aschenaziti addirittura del 97%. Nelle popolazioni del Sud Europa e in particolare in Italia queste percentuali si riducono molto. In Italia vi sono notevoli differenze regionali nello spettro di mutazioni FC. Conseguentemente, nonostante siano attualmente utilizzati kit diagnostici per 32 mutazioni, ma che offrono la possibilità di ricercare sino a 57 mutazioni, comprese le mutazioni più frequenti nella popolazione italiana, vi è una grande variabilità di efficienza diagnostica che va dal 92% della Sardegna al 67% della Lombardia. Nella tab. 4 viene riportata la proporzione di mutazioni identificabili e il rischio residuo di avere un figlio con FC per la coppia portatore-non portatore.
Sono stati proposti due approcci allo screening della coppia: il primo studia entrambi i componenti contemporaneamente e valuta in seguito ai risultati ottenuti un rischio residuo di coppia di generare figli affetti. Nel secondo, chiamato sequenziale, si studia prima un componente della coppia, generalmente la donna, e solo in caso dovesse risultare portatore si analizza anche il partner, con lo stesso pannello di mutazioni o possibilmente con un pannello più ampio. La consulenza genetica acquista, nei programmi di prevenzione della fibrosi cistica, una importanza fondamentale poiché permette di chiarire il concetto che risultare negativi allo screening di coppia significa abbassare fortemente il rischio di generare un figlio affetto ma non di escluderlo e nel contempo consente di non ingenerare ansie ingiustificate.
Screening fetali
Lo screening genetico di donne apparentemente sane in corso di gravidanza è quel procedimento che tende a identificare difetti congeniti in coppie senza alcun rischio a priori. Questo tipo di screening si articola essenzialmente su tre procedimenti diagnostici: (a) determinazione della compatibilità Rh; (b) valutazione del feto tramite analisi con ultrasuoni; (c) analisi biochimiche sul sangue materno con lo scopo di identificare feti a rischio per aberrazioni cromosomiche e per difetti del tubo neurale. Ovviamente lo screening deve essere preceduto da appropriata consultazione genetica.
Determinazione della compatibilità Rh
La determinazione della compatibilità Rh madre-feto ha lo scopo di prevenire la malattia emolitica del neonato dovuta a incompatibilità in questo sistema. La malattia emolitica da isoanticorpi si verifica quando la madre è Rh negativa e il feto Rh positivo. In queste condizioni, gli eritrociti fetali, che in numero limitato anche di norma attraversano la placenta e raggiungono il circolo materno, stimolano la produzione di anticorpi anti-Rh. L'immunizzazione della madre può avvenire anche con altre modalità, come vedremo successivamente. Questi anticorpi anti-Rh attraversano la placenta, raggiungono il feto e provocano emolisi dei globuli rossi. Ne deriva una grave anemia. A seconda della gravità vengono distinti tre quadri clinici: l'anemia emolitica del neonato, l'ittero grave del neonato e l'idrope feto-placentare. Quest'ultima è il quadro più grave ed è dovuta essenzialmente a una insufficienza cardiaca secondaria all'anemia fetale. L'incidenza di questa malattia varia ovviamente in funzione della prevalenza nella popolazione di soggetti Rh negativi. Questa differisce notevolmente da popolazione a popolazione.
La Rh positività comprende individui eterozigoti che hanno ereditato il gene D da un solo genitore e individui omozigoti che hanno ereditato il gene D da entrambi i genitori. Il genotipo Rh D negativo è dovuto ad assenza totale o parziale del gene Rh D. L'immunizzazione di una madre Rh negativa può avvenire tramite una trasfusione di sangue Rh positivo o per il passaggio dei globuli rossi fetali nel circolo materno durante o a termine di una gravidanza. Quest'ultima è l'evenienza più comune e spiega come la malattia emolitica da incompatibilità Rh sia eccezionale nella prima gravidanza. L'immunizzazione Rh si verifica più di frequente quando la gravidanza è complicata da tossiemia, parto cesareo o rimozione manuale della placenta, tutte condizioni che facilitano una emorragia transplacentare. L'emorragia feto-placentare avviene in modo predominante al momento del parto o di un aborto, oppure dopo manovre diagnostiche quali la villocentesi, l'amniocentesi o la fetoscopia. Ovviamente l'immunizzazione è tanto più frequente e più seria quanto maggiore è stata l'emorragia transplacentare.
La prevenzione della malattia emolitica del neonato da incompatibilità Rh si fonda sul trattamento della donna Rh negativa con partner Rh positivo con anticorpi anti-Rh subito dopo il parto, l'aborto o una manovra diagnostica. La dose di immunoglobuline anti-Rh dovrebbe essere stabilita tramite una valutazione del numero di globuli rossi fetali presenti nel sangue materno. Nell'1-2% delle donne Rh negative l'immunizzazione avviene prima del parto. In questa situazione la comparsa di anticorpi si verifica per lo più dopo la 28a settimana di gravidanza. La prevenzione di questi casi può essere ottenuta facendo la profilassi con anticorpi anti-Rh alla 28a settimana. Questo trattamento non mostra effetti negativi sul feto. In caso la donna in gravidanza sia già stata immunizzata, la conoscenza di questo stato di incompatibilità del feto in utero può consentire di evitare la morte applicando trattamenti appropriati.
Ecografia del feto
L'analisi con ultrasuoni è attualmente il sistema migliore per la individuazione prenatale di anomalie congenite. L'ecografia del feto consente di svelare da un lato malformazioni maggiori, ma permette anche di identificare alterazioni minori indicative di anomalie cromosomiche o di altri difetti genetici. Attualmente viene raccomandato che ogni donna gravida sia sottoposta almeno a due ecografie fetali, una alla 11-13a, utile per la datazione della gravidanza e per lo studio della translucenza nucale, e una alla 20a settimana di gravidanza per valutare l'accrescimento del feto e per una analisi sistematica dei difetti maggiori e minori. Tra le più importanti anomalie congenite e i difetti genetici identificabili con l'ecografia fetale, per la loro frequenza vanno menzionati in particolare i difetti del tubo neurale (anencefalia e spina bifida), idrocefalia, oloprosencefalia, macrocefalia, malformazione cistica adenomatosa del polmone, ernia diaframmatica, idrope fetale, difetti della parete addominale, difetti gastrointestinali, cardiaci e renali, molte displasie scheletriche, oligo- e poliidramnios che possono essere marcatori di anomalie fetali.
Oltre queste anomalie maggiori vi sono una serie di alterazioni minime che indicano la possibile presenza di anomalie cromosomiche. Tra queste riveste particolare rilievo il riscontro di igroma cistico o la misura della translucenza nucale (NT) intesa come lo spessore della regione posteriore del collo compresa tra cute e colonna vertebrale, o l'accorciamento relativo del femore e il ritardo di accrescimento intrauterino. In feti con alterazioni all'ecografia l'incidenza di anomalie cromosomiche è elevata, pari a circa il 2% in presenza di un difetto singolo e fino al 27% quando vi è più di una malformazione. Considerando il problema da un altro punto di vista si può affermare che il 90% dei feti con trisomia 13, 18, di quelli triploidi o con sindrome di Turner hanno malformazioni associate che sono evidenziabili tramite un accurato esame ecografico fetale. Nella trisomia 21, invece, le alterazioni fetali sono più sottili e la diagnosi ecografica più difficile. Negli ultimi anni, comunque, una attenta valutazione della translucenza nucale, eseguita in centri specializzati, si è rivelata un attendibile metodo di screening precoce.
Screening per la sindrome di Down
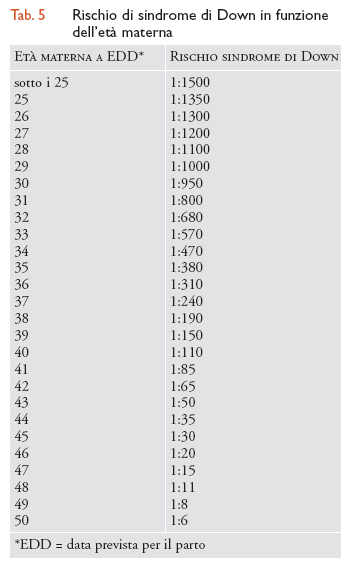
La sindrome di Down (o trisomia del cromosoma 21) è la più comune causa di grave insufficienza mentale. Oltre l'insufficienza mentale, i bambini con sindrome di Down hanno diverse anomalie tra cui taglio obliquo degli occhi, cute sovrabbondante sulla nuca, macroglossia, naso piatto, clinodattilia, solco unico delle quattro dita e incostantemente alcune malformazioni maggiori, tra cui più frequenti quelle cardiache. La sua incidenza è di 1:700 nati vivi. L'insufficienza mentale è di varia entità, ma per lo più è grave. Alcuni individui con trisomia 21 hanno vita serena e indipendente, altri sono completamente dipendenti per le loro attività. Il 50% delle gravidanze con sindrome di Down esitano in aborti. Circa 1/3 dei casi hanno un difetto cardiaco serio. La previsione di vita è attualmente di 60 anni circa. Molti casi sviluppano un quadro tipo Alzheimer, cioè una demenza progressiva dopo i 40 anni. L'unico fattore di rischio è l'età materna. Come risulta evidente dall'esame della tab. 5, il rischio aumenta da 1:1500 sotto i 25 anni a 1:6 sopra i 50 anni. Alle donne con età superiore ai 35-38 anni viene proposta la diagnosi prenatale invasiva che consiste nell'analisi del cariotipo fetale (ricerca di anomalie cromosomiche), su cellule del liquido amniotico prelevate mediante amniocentesi (alla 16a-17a settimana di gravidanza) o su cellule del trofoblasto prelevate mediante villocentesi, in epoca gestazionale molto più precoce, alla 11a settimana. Entrambe le tecniche di prelievo hanno un rischio di perdita fetale dell'1% circa, se eseguite da personale specializzato.
A queste indagini sfuggono circa il 50% dei casi che avvengono in gravidanze non definite a rischio, cioè in donne con età inferiore a 35 anni. Per questa ragione sono stati sviluppati negli ultimi anni dei protocolli di screening biochimici e ultrasonografici, che hanno come target tutte le gravidanze indipendentemente da fattori di rischio aggiuntivi e che combinati insieme ed eseguiti in epoca gestazionale appropriata hanno un potere di predittività molto elevato e una bassa percentuale di falsi positivi. Lo schema dello screening può essere riassunto come segue.
Test combinato, ecografico e sierologico. - Si attua tra la 11a e la 13a settimana di gravidanza e comprende: (a) valutazione ecografia della translucenza nucale (NT); (b) misura dei livelli plasmatici materni di β-hCG (subunità libera β delle gonadotropine corioniche) e della proteina plasmatica A associata alla gravidanza (PAPP-A) correlati con l'età materna. È stato osservato che usando entrambe le valutazioni piuttosto che la NT da sola il potere predittivo aumenta dal 70 all'87%, con un rischio di avere falsi positivi del 5%. Il test combinato fornisce i risultati più attendibili se eseguito alla 11a settimana di gravidanza.
Test sierologico. - Si effettua alla 15a-18a settimana di gravidanza e consiste nel dosaggio sierico di: α-fetoproteina, hCG totali (doppio test); α-fetoproteina, hCG totali, estriolo non coniugato (triplo test); α-fetoproteina, hCG totali, estriolo non coniugato, inibina A (quadruplo test).
Questi test, congiuntamente con la valutazione dell'età materna, consentono di eseguire una valutazione quantitativa del rischio. Nella gravidanza con sindrome di Down, l'α-fetoproteina e l'estriolo coniugato sono ridotti, le hCG e l'inibina A sono aumentate. Sulla base dei valori di questi esami e dell'età materna viene fatto un calcolo probabilistico; con un rischio di 1/150 al primo trimestre e di 1:300 al secondo trimestre il test viene giudicato positivo e viene discussa l'opzione per ulteriori analisi.
Il test sierologico al secondo trimestre sembrerebbe avere una più alta percentuale di falsi positivi del test combinato (NT, β-hCG, PAPP-A) al primo trimestre, sempre che questo sia eseguito alla 11a settimana di gravidanza e la misura ecografica della translucenza nucale sia valutata da personale altamente specializzato. Il test sierologico integrato (test sierologico al primo e al secondo trimestre) ha risultati simili a quelli del test combinato alla 11a settimana di gravidanza e può essere utilizzato laddove la valutazione della NT non sia disponibile. L'associazione del test combinato al primo trimestre con il quadruplo test nel secondo trimestre viene chiamata 'test integrato' ed è quello che presenta migliore potere predittivo (96%) e migliore specificità. In caso di positività (rischio > 1/150-300), la diagnosi definitiva può essere ottenuta solamente con l'indagine del cariotipo attraverso amniocentesi o villocentesi. È chiaro che in caso di risultato positivo al test combinato al primo trimestre, l'orientamento è quello di proporre subito una indagine invasiva da effettuarsi nel primo trimestre attraverso villocentesi, mentre il test integrato dovrebbe essere proposto nelle gravidanze risultate negative al test combinato.
In molti Paesi sono state intraprese iniziative per estendere a tutte le donne, indipendentemente dall'età, il test di screening per la sindrome di Down, ponendo però come condizione imprescindibile che vengano raggiunti degli obiettivi concreti di miglioramento degli standard di efficacia e sicurezza dei test. Occorre infatti migliorare la efficienza diagnostica del test, aumentando il numero donne con feto affetto rilevate, ma contemporaneamente diminuendo i casi di falsa positività che portano ad avere un eccesso di interventi di diagnosi prenatale, con conseguenti inutili rischi aggiuntivi per il feto.
Screening fetale dei difetti del tubo neurale
I difetti del tubo neurale sono dovuti a difettosa chiusura delle docce neurali primitive e comprendono l'anencefalia e i vari tipi di spina bifida (mielomeningocele, meningocele e spina bifida occulta). Essi sono tra i più comuni e gravi difetti congeniti. La loro origine è polifattoriale, includendo fattori ambientali e genetici. In assenza di screening prenatale l'incidenza è di 1:400 bambini nati vivi. Tra i difetti del tubo neurale l'anencefalia è fatale nel periodo neonatale. La storia naturale della spina bifida è variabile. Per lo più i bambini con spina bifida hanno paresi degli arti inferiori, incontinenza fecale e urinaria, idrocefalo e meno frequentemente insufficienza mentale. I difetti del tubo neurale si verificano per una alterazione del processo di chiusura dell'abbozzo primitivo del midollo, che da una fase primitiva di placca si trasforma successivamente in doccia e infine in canale chiuso.
I difetti del tubo neurale sono diagnosticabili mediante valutazione ecografica al secondo trimestre di gravidanza e mediante dosaggio dell'AFP, compresa nei test biochimici precedentemente descritti, che risulta aumentata in queste condizioni. Le donne che mostrano un valore pari a 2,5 volte la media vengono considerate positive. Segue la consultazione genetica, durante la quale vengono discusse le opzioni per ulteriori esami. Una donna su 50 risulta positiva all'esame di screening. Di queste donne 1:20 ha una gravidanza con feto affetto. Nel complesso vengono identificate 2 donne su 3 con feto affetto da difetto del tubo neurale e praticamente tutte le gravidanze con anencefalia.
Bibliografia
Botkin 2005: Botkin, Jeffrey R., Research for newborn screening: developing a national framework, "Pediatrics", 116, 2005, pp. 862-871.
Cao 1993: Cao, Antonio - Galanello, Renzo - Rosatelli, Maria Cristina, Screening and prenatal diagnosis of the haemoglobinopathies, "Baillière's clinical haematology", 6, 1993, pp. 263-286.
Godard 2003: Godard, Béatrice e altri, Population genetic screening programmes: principles, techniques, practices, and policies, "European journal of human genetics. Suppl.", 2, 2003, S49-S87.
Grody 2001: Grody, Wayne W. e altri, Laboratory standards and guidelines for population-based cystic fibrosis carrier screening, "Genetics in medicine", 3, 2001, pp. 149-154.
Kaback 1993: Kaback, Michael e altri, Tay-Sachs disease. Carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1979 to 1993, "Journal of the American Medical Association", 270,1993, pp. 2307-2315.
Khoury 2003: Khoury, Muin J. - McCabe, Linda L. - McCabe, Edward R., Population screening in the age of genomic medicine, "New England journal of medicine", 348, 2003, pp. 50-58.
Malone 2005: Malone, Fergal D. e altri, First- and second-trimester evaluation of risk (FASTER) research consortium. First-trimester or second-trimester screening, or both, for Down's syndrome, "New England journal of medicine", 353, 2005, pp. 2001-2011.
Milunsky 1992: Genetic disorders and the fetus: diagnosis, prevention and treatment, 3. ed., edited by Aubrey Milunsky, Baltimore, John Hopkins University Press, 1992.
Natowicz 2005: Natowicz, Martin, Newborn screening - setting evidence-based policy for protection, "New England journal of medicine", 353, 2005, pp. 867-870.
Petersen, Codori 2001: Petersen, Gloria M. - Codori Ann-Marie, Genetic testing for familial cancer, in: The metabolic & molecular bases of inherited disease, edited by Charles R. Scriver e altri, New York, McGraw-Hill, 2001, pp. 1063-1076.
Wald 2003: Wald, Nicholas J. e altri, First and second trimester antenatal screening for Down's syndrome: the results of the serum, urine and ultrasound screening study (SURUSS), "Health technology assessment", 7, 2003, pp. 1-88.
Watson 2001: Watson, Michael S. e altri, Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel, "Genetics in medicine", 3, 2001, pp. 149-154.
Wilcken 2003: Wilcken, Bridget e altri, Screening newborns for inborn errors of metabolism by tandem mass spectrometry, "New England journal of medicine", 348, 2003, pp. 2304-2312.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


