Proteine
Enciclopedia del Novecento (1980)
Proteine
Metodi di studio e struttura covalente, di G. Michael Hass e Hans Neurath
Struttura tridimensionale, di Giovanni Ronca
Metodi di studio e struttura covalente
SOMMARIO: 1. Introduzione. □ 2. Amminoacidi. □ 3. Legame covalente. □ 4. Caratterizzazione fisica e chimica delle proteine: a) purificazione; b) peso molecolare; c) composizione; d) gruppi terminali. □ 5. Determinazione della sequenza degli amminoacidi: a) tecniche sperimentali; b) verifica delle sequenze determinate chimicamente; c) informazioni ricavate dallo studio della sequenza. □ 6. Fattori che influenzano la conformazione proteica. □ 7. Metodi per determinare la struttura tridimensionale. □ 8. Considerazioni conclusive. □ Bibliografia.
1. Introduzione.
Le proteine (dal greco πρώτειος principale, primario) si trovano in tutte le forme note di vita; sono macromolecole il cui peso molecolare può variare tra 5.000 circa e diversi milioni. Una molecola proteica è formata da una o più catene polipeptidiche, composte di α-amminoacidi legati insieme da legami peptidici (v. sotto); il modo in cui le catene sono ripiegate determina le possibilità funzionali di ciascuna proteina.
Al principio del Novecento non si conosceva praticamente nulla circa la chimica delle proteine: la nostra conoscenza della struttura di queste molecole è una delle principali acquisizioni degli scienziati del XX secolo. La necessità di isolare queste sostanze così labili da una miscela di molecole simili e la mancanza di una tecnologia adeguata per studiare le proteine una volta isolate sono state le maggiori difficoltà da superare; le soluzioni a molti di questi problemi sono state ottenute applicando e modificando tecniche proprie della chimica fisica, analitica e organica. Inoltre è stata sviluppata una tecnologia interamente nuova, specifica per affrontare i problemi che si presentano al chimico delle proteine: per esempio le tecniche della cromatografia a scambio ionico, dell'elettroforesi, della gelfiltrazione, dell'ultracentrifugazione analitica e diverse altre sono state sviluppate primariamente per la purificazione e la caratterizzazione delle proteine.
Oltre a mancare di una tecnologia adeguata, il biochimico all'inizio del XX secolo era sotto l'influenza del dogma secondo il quale le proteine non erano entità discrete, ma piuttosto strutture labilmente organizzate, di composizione variabile. Questo dogma è stato forse altrettanto nocivo per lo sviluppo della biochimica delle proteine quanto il vitalismo per lo sviluppo della chimica organica nel secolo precedente. La dimostrazione che le proteine sono molecole chimicamente e fisicamente omogenee ha costituito il fondamento concettuale per lo studio della struttura proteica. Le dimostrazioni di omogeneità erano basate: 1) sulla cristallizzabilità; 2) sulla non variabilità nella composizione e nella sequenza degli amminoacidi (v. sotto); 3) sul comportamento simile a quello di sistemi monodispersi durante la sedimentazione; 4) sulla specificità della struttura tridimensionale di alcune proteine.
Un discorso sulla struttura delle proteine può essere affrontato ponendo le seguenti domande: 1) quali sono gli amminoacidi costitutivi delle proteine? 2) qual è la sequenza di questi amminoacidi e quale informazione vi è contenuta? 3) qual è la struttura tridimensionale delle proteine e quali forze la determinano e la stabilizzano?
2. Amminoacidi.
Le proteine sono polimeri di α-amminoacidi; questi ultimi hanno la formula generale
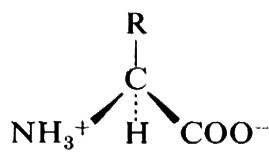
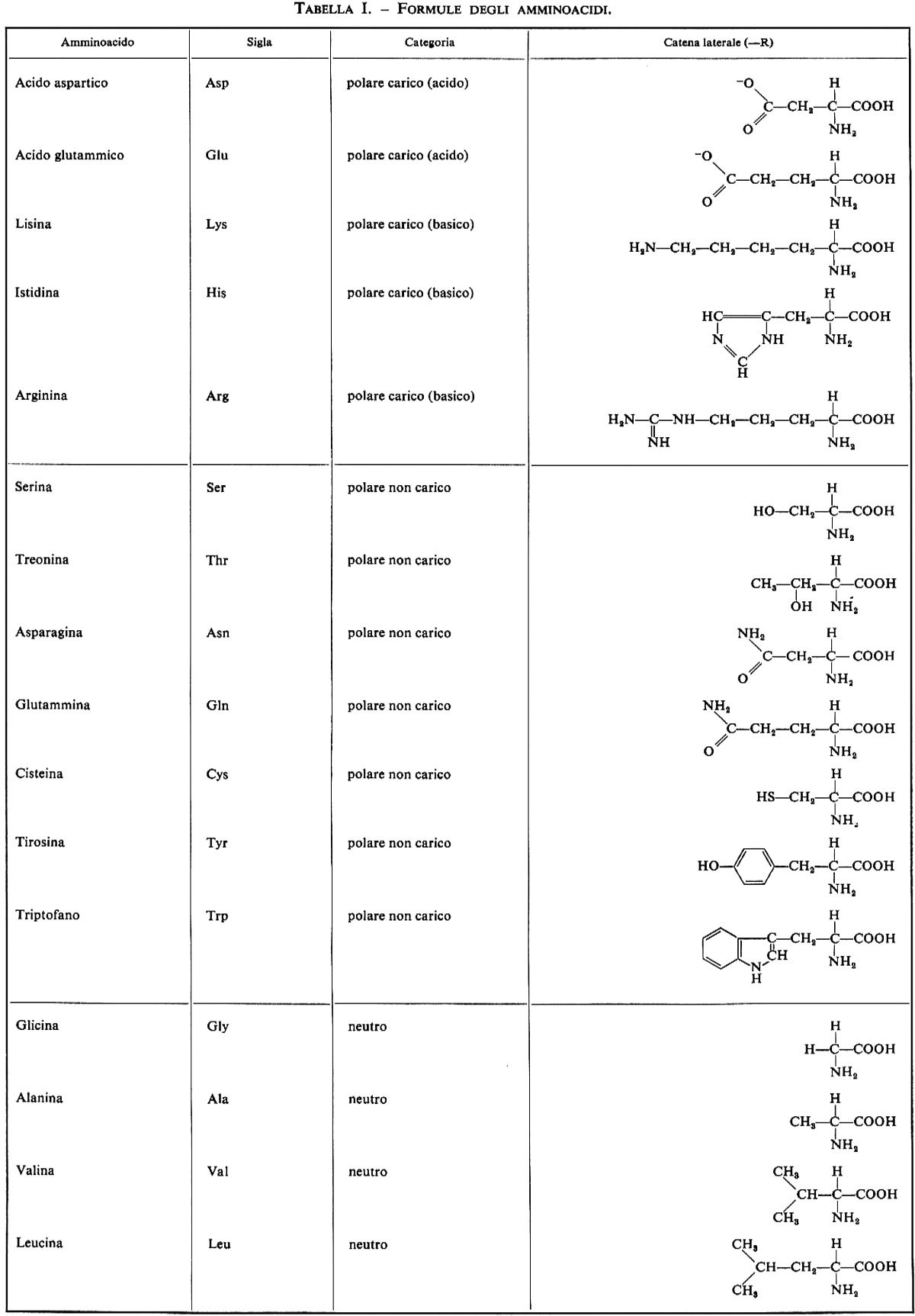
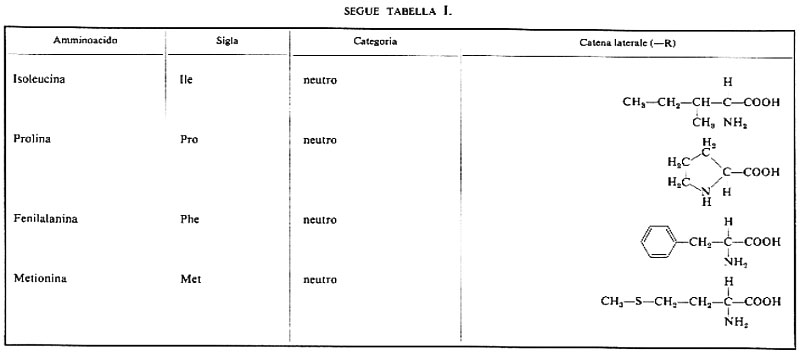
Dei numerosissimi composti che possono avere questa formula, solo 20 si trovano comunemente nelle proteine; le formule di questi amminoacidi e della prolina, che a rigor di termini è un imminoacido, sono riportate nella tab. I. La possibilità di trovare 20 amminoacidi diversi in ogni posizione lungo una catena polipeptidica è causa della enorme diversità nella struttura delle proteine: ammesso che ciascun amminoacido possa trovarsi in una qualsiasi posizione all'interno di una proteina la cui molecola sia formata da 100 molecole di amminoacidi, si potrebbero avere 20100 specie di proteine diverse; è possibile calcolare che se ciascuna di queste specie fosse rappresentata anche da una sola molecola, la somma totale sarebbe superiore a quella che rende conto di tutta la massa nell'Universo.
Nelle proteine sono stati trovati sempre solo amminoacidi della configurazione L, dove L si riferisce alla configurazione assoluta e non all'attività ottica (v. stereochimica).

Gli isomeri D si trovano in alcune strutture come la parete della cellula batterica e particolari antibiotici; resta oscuro il motivo dell'assenza dei D-amminoacidi nelle proteine.
La glicina non ha atomi di carbonio asimmetrici e quindi non è né D né L; gli amminoacidi isoleucina, treonina e cistina hanno due centri di asimmetria, quindi vi sono quattro isomeri possibili per ognuno di essi, salvo la cistina che, avendo due carboni asimmetrici equivalenti, può dar luogo solo a tre isomeri: L-cistina, D-cistina e meso-cistina.
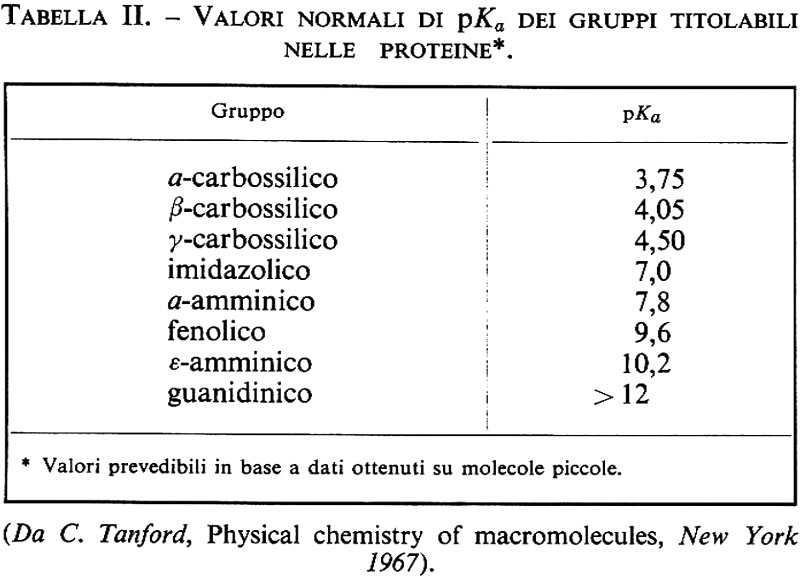
Come si vede dalla tab. I, la natura della catena laterale R varia molto nei diversi amminoacidi; questi sono in genere classificati, a seconda della natura della catena laterale, in polari carichi, polari non carichi, e non polari o neutri: i primi hanno nella catena laterale un gruppo ionizzabile e si dividono in basici (lisina, istidina, arginina) e acidi (acido aspartico e acido glutammico); benché il gruppo fenolico della tirosina e il gruppo solfidrilico della cisteina possano ionizzarsi, questi due amminoacidi vengono classificati tra i polari non carichi a causa delle loro caratteristiche complessive. La presenza di questi gruppi ionizzabili rende le proteine polielettroliti la cui carica varia in funzione del pH: i valori normali di pKa di questi gruppi sono riportati nella tab. II.
La seconda categoria, costituita dagli amminoacidi polari non carichi, che hanno la catena laterale non ionizzabile o pochissimo ionizzabile, comprende serina, treonina, cisteina, asparagina, glutammina, triptofano e tirosina; questi amminoacidi possono formare legami a idrogeno.
La terza categoria, amminoacidi non polari o neutri, comprende glicina, alanina, valina, leucina, isoleucina, prolina, metionina e fenilalanina. Quando questi amminoacidi sono sciolti in acqua, le molecole di solvente intorno alle catene laterali tendono a formare strutture ordinate. Le interazioni fra parecchie di tali catene laterali causano una disorganizzazione dell'acqua e, di conseguenza, un favorevole aumento di entropia del sistema; questo tipo di interazione, detta idrofobica, è ritenuto il principale responsabile della formazione e del mantenimento della struttura proteica. L'importanza relativa delle diverse interazioni tra le catene laterali nel determinare la struttura tridimensionale di una proteina sarà discussa in seguito.
3. Legame covalente.
I due principali legami che si riscontrano nelle proteine sono il legame peptidico e il ponte disolfuro. Nel 1902 fu avanzata l'ipotesi che gli amminoacidi fossero uniti con legame peptidico (v. Fischer, 1902; v. Hofmeister, 1902). Quest'ipotesi era basata sulle seguenti osservazioni: 1) benché le proteine come tali contengano pochi gruppi amminici e carbossilici liberi, la loro idrolisi chimica o enzimatica libera quantità eguali di questi gruppi; 2) enzimi capaci di idrolizzare le proteine sono capaci di idrolizzare anche piccoli substrati peptidici sintetici; 3) l'idrolisi parziale delle proteine produce di- e tripeptidi identici a quelli ottenuti per sintesi. Il legame peptidico è formato per condensazione del gruppo α-amminico di un amminoacido col gruppo carbossilico dell'amminoacido adiacente: la catena polipeptidica è un polimero formato da amminoacidi uniti in questo modo.
Le distanze interatomiche e gli angoli di legame sono stati determinati con la cristallografia a raggi X in diversi amminoacidi e piccoli peptidi allo stato cristallino (v. Pauling e altri, 1951): tali valori sono indicati nella fig. 1 per un segmento peptidico.
La fig. 1 riporta la forma trans del legame peptidico; la forma cis è molto rara. Di particolare interesse è la lunghezza di 1,32 Å del legame C−N; questo valore è intermedio tra quello tipico del legame singolo carbonio-azoto (≈1,49 Å) e quello del doppio legame carbonio-azoto (1,27 Å); ciò indica che il legame peptidico ha notevoli caratteri di doppio legame e che si tratta in realtà di un ibrido di risonanza. Tale delocalizzazione di elettroni intorno all'azoto, al carbonio e all'ossigeno ammidici produce un massimo di stabilità quando gli atomi suindicati sono coplanari; la coplanarità di questi atomi riduce grandemente le possibilità di flessione della catena polipeptidica: di queste restrizioni si parlerà più avanti.
Lo studio dell'idrolisi del composto modello N-benzoil-glicil-tirosinammide indica che l'energia libera di idrolisi del legame peptidico è circa −0,4 kcal/mole; poiché è facile idrolizzare le proteine ad amminoacidi, era prevedibile che questa energia libera fosse negativa. Da questa osservazione si traggono due conseguenze: 1) le proteine sono (relativamente) instabili in condizioni fisiologiche e possono essere degradate in presenza di enzimi idrolitici; da questo punto di vista si comportano come altri polimeri biologici (acidi nucleici, carboidrati), che hanno anch'essi un valore negativo di energia libera di idrolisi; 2) la sintesi delle proteine richiede energia (v. Watson, 1965).
Mentre il legame peptidico è il mezzo per unire i vari amminoacidi a formare la catena polipeptidica, il ponte disolfuro tra due semiresidui di cistina serve a stabilizzare la struttura proteica; ciò viene realizzato congiungendo o differenti regioni della stessa catena polipeptidica (legame intracatena) o due differenti catene polipeptidiche (legame intercatene). Per esempio l'insulina (v. fig. 2) contiene sia un ponte disolfuro intracatena (Cys-A6→Cys-A11) sia due ponti intercatene (Cys-A7→Cys-B7; Cys-A20→Cys-B19).
Il legame disolfuro può essere ridotto per trattamento con composti tiolici in eccesso (per es. 2-mercaptoetanolo) o con boroidruro di sodio, con produzione di residui di cisteina (−SH); la reazione col 2-mercaptoetanolo è la seguente:
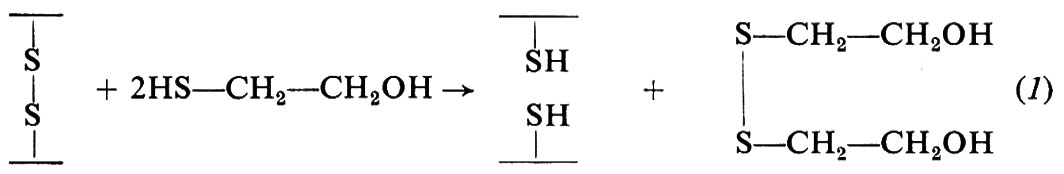
Il ponte disolfuro può essere anche aperto mediante ossidazione con acido performico (2) o trattamento con solfito (3); queste reazioni vengono usate per l'analisi strutturale delle proteine:
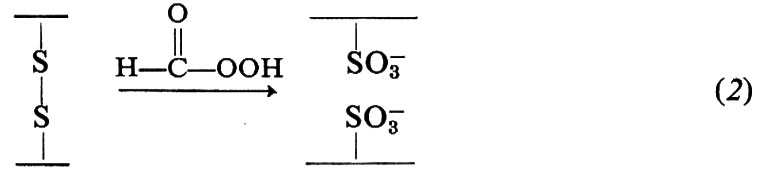
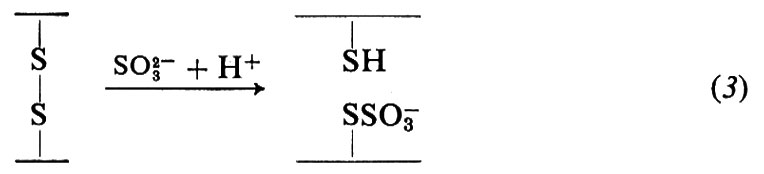
La presenza di ponti disolfuro in una proteina limita notevolmente le possibilità conformazionali di una catena di amminoacidi, così come la coplanarità richiesta dal legame peptidico limita la flessibilità di un polipeptide; tuttavia rimangono possibili numerose conformazioni.
4. Caratterizzazione fisica e chimica delle proteme.
Uno studio completo delle proprietà strutturali di una proteina comprende: 1) la sua caratterizzazione generale; 2) l'analisi della sequenza degli amminoacidi; 3) la determinazione della conformazione della proteina. Questi studi richiedono spesso quantità relativamente grandi di proteina purificata (v. enzimi).
a) Purificazione.
La proteina studiata è spesso presente in piccole quantità (〈1%) in una miscela complessa di altre proteine con caratteristiche fisiche simili. Complica il problema il fatto che la purificazione dev'essere effettuata in condizioni blande, poiché valori estremi di temperatura, di pH e composizione del solvente spesso causano l'irreversibile denaturazione della proteina con conseguente perdita dell'attività biologica. Tutte queste difficoltà hanno portato allo sviluppo di tecniche di purificazione molto selettive. I primi metodi di purificazione si basavano sulla differente solubilità delle proteine in varie soluzioni saline e in tamponi contenenti solventi organici; più recentemente sono stati messi a punto sistemi cromatografici che separano in base a una differenza di carica (scambio ionico), di dimensioni (gelfiltrazione), di specifici legami (cromatografia di affinità); una proteina può essere generalmente purificata usando una combinazione di alcuni di questi metodi. Il criterio usuale di purezza, per una proteina, è l'omogeneità, determinata con metodi che separano le proteine in base alla carica (per es. elettroforesi, focalizzazione isoelettrica), alla dimensione (velocità di sedimentazione, elettroforesi su gel in presenza di dodecilsolfato di sodio) e a caratteristiche immunologiche (metodo di O. Ouchterlony).
b) Peso molecolare.
Come accennato in precedenza, il peso molecolare di una proteina può variare tra circa 5.000 e parecchi milioni; poiché il peso molecolare medio per residuo è 110, le proteine possono contenere da circa 45 a molte migliaia di residui di amminoacidi per molecola; catene formate da meno di 50 residui circa di amminoacidi sono comunemente indicate col nome di peptidi, ma si tratta di una distinzione puramente semantica.
Vi sono numerosi metodi per la determinazione del peso molecolare delle proteine. Ricordiamo quelli che utilizzano: 1) la diffusione della luce; 2) la pressione osmotica; 3) la gelfiltrazione; 4) la microscopia elettronica; 5) l'elettroforesi su gel di poliacrilammide; 6) l'equilibrio di sedimentazione: quest'ultimo è di particolare importanza.
Nel 1923 fu costruita l'ultracentrifuga allo scopo di determinare il peso molecolare di macromolecole (v. Svedberg e Pederson, 1940); in essa il rotore, contenente una cella con la soluzione proteica, viene fatto girare a velocità relativamente bassa in modo che la forza di sedimentazione venga esattamente bilanciata dalla forza di diffusione; all'equilibrio si avrà:
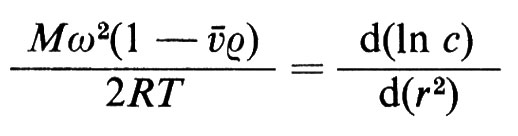
in cui M è il peso molecolare, ω la velocità angolare, ã il volume specifico parziale della proteina, ρ la densità del solvente, c la concentrazione della proteina ed r la distanza dall'asse di rotazione. Usando un'ottica interferenziale si ottiene la concentrazione c della proteina in funzione di r: se il grafico di ln c in funzione di r2 è lineare, dalla pendenza della retta ottenuta si può calcolare il peso molecolare; una non linearità suggerisce un'eterogeneità della preparazione o la presenza di fenomeni di associazione-dissociazione.
Benché le proteine possano avere pesi molecolari di diversi milioni, le proteine più grandi sono in genere composte da subunità polipeptidiche più piccole; queste subunità hanno spesso un peso molecolare inferiore a 100.000 e sono tenute insieme da interazioni non covalenti o da ponti disolfuro (v. tab. III); trattando queste proteine con agenti denaturanti, come urea, sali di guanidina o dodecilsolfato di sodio (un detergente anionico), si eliminano le interazioni non covalenti e si separano le subunità; in presenza di agenti riducenti si aprono anche i ponti disolfuro.
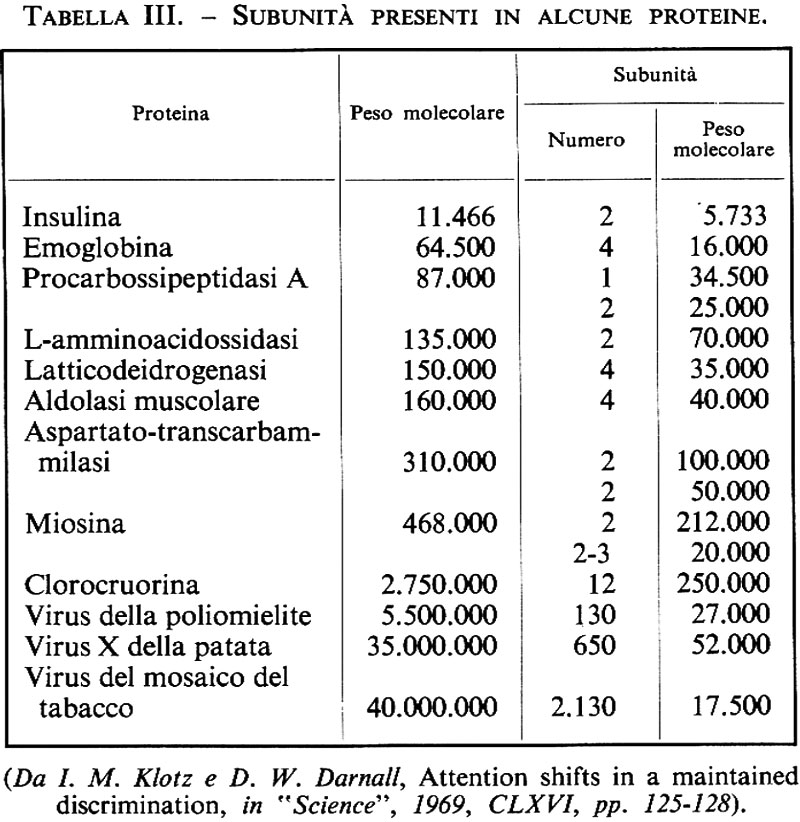
Il peso molecolare delle subunità viene determinato molto accuratamente (±5%) con l'equilibrio di sedimentazione in solventi denaturanti contenenti sostanze riducenti; un metodo alquanto meno accurato ma rapido, particolarmente adatto nel caso di subunità diseguali, è rappresentato dall'elettroforesi su gel in presenza di dodecilsolfato di sodio. I rapporti tra logaritmo del peso molecolare della subunità e mobilità elettroforetica, per diverse proteine, sono riportati nella fig. 3: il confronto della mobilità elettroforetica di una subunità a peso molecolare ignoto con quelle di altre note fornisce prontamente, con ragionevole approssimazione, il peso molecolare della subunità stessa.
c) Composizione.
Le quantità relative di ciascun amminoacido in una proteina possono essere determinate dopo idrolisi completa della stessa; si è trovato che l'idrolisi effettuata in HCl 6 N a 110° sotto vuoto per 24 ore produce una scissione pressoché completa della proteina con minima distruzione di amminoacidi, eccetto il triptofano, l'asparagina e la glutammina; la cisteina e la cistina si determinano meglio sotto forma di acido cisteico dopo ossidazione con acido performico (v. eq. 2). Oggi si adoperano largamente metodi automatici per l'analisi cromatografica degli amminoacidi: questi sono separati su di una resina polistirenica solfonata e determinati quantitativamente facendo reagire l'eluato con ninidrina, per formare un composto colorato: l'intensità del colore (assorbanza o estinzione) viene misurata e registrata automaticamente, e ne risulta un diagramma come quello riportato in fig. 4; l'area al di sotto di ogni picco è proporzionale alla quantità di amminoacido presente.
d) Gruppi terminali.
La determinazione qualitativa e quantitativa degli amminoacidi ammino- e carbossi-terminali di una proteina non solo fornisce un'ulteriore informazione strutturale, ma può anche essere usata per determinare il numero di catene polipeptidiche che formano la proteina e per accertarne l'omogeneità: per esempio, la presenza di 2 moli di alanina ammino-terminale per mole di proteina indica che la proteina è un dimero, mentre il trovare 0,8 moli di alanina e 0,2 di leucina per mole di proteina indica una preparazione eterogenea. Il metodo migliore per determinare i residui ammino-terminali è quello del cianato, che reagisce a pH alcalino con i gruppi α-amminici delle proteine nel modo seguente:
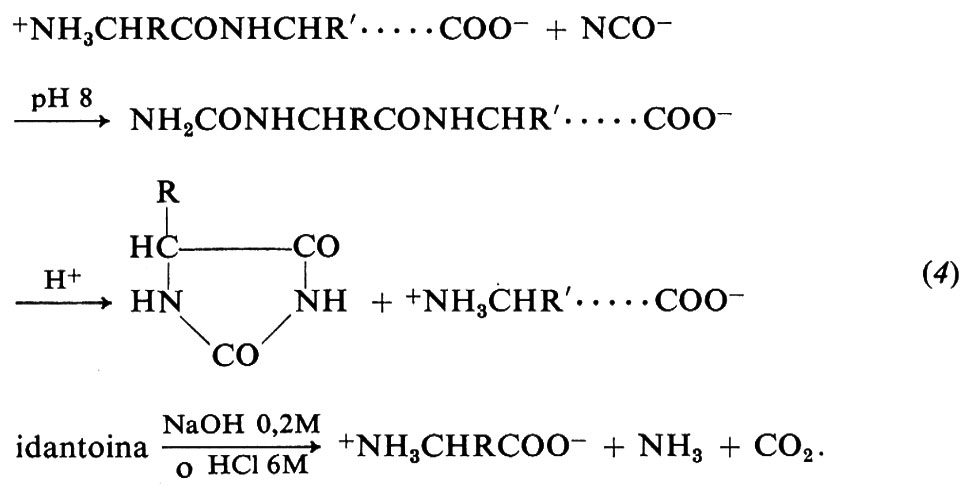
Come si vede dall'eq. (4), il trattamento con acido produce il distacco dell'amminoacido ammino-terminale e la sua ciclizzazione a idantoina; quest'ultima viene isolata cr0matograficamente e idrolizzata con acidi o basi per liberare di nuovo detto amminoacido.
L'amminoacido carbossi-terminale viene generalmente identificato tramite l'idrazinolisi o la digestione della proteina con carbossipeptidasi. L'idrazina trasforma tutti gli amminoacidi della catena polipeptidica, eccetto il residuo carbossi-terminale, nelle idrazidi corrispondenti, le quali possono essere separate cromatograficamente dall'amminoacido carbossi-terminale non modificato. Le carbossipeptidasi staccano sequenzialmente gli amminoacidi a partire dall'estremità carbossilica della catena polipeptidica: quando una proteina dalla sequenza H2N−Leu-Glu-PheCOOH viene trattata con carbossipeptidasi A e si - fanno prelievi nel tempo per seguire la comparsa di amminoacidi liberi, si ottiene un risultato come quello riportato in fig. 5.
5. Determinazione della sequenza degli amminoacidi.
Nel 1955 fu pubblicata la prima sequenza degli amminoacidi di una proteina a molecola relativamente piccola, l'ormone insulina (v. Sanger e altri, 1955): questo risultato è stato veramente una pietra miliare nello studio della struttura delle proteine, tanto più notevole se si considerano le tecniche relativamente primitive disponibili a quel tempo. Questo studio non solo ha dimostrato che le proteine hanno struttura chimica definita, ma ha ulteriormente stimolato la ricerca in questo campo: da allora la tecnologia per l'analisi della sequenza degli amminoacidi è stata molto perfezionata e sono state così determinate le sequenze di centinaia di catene polipeptidiche. Queste sequenze sono riportate in una pubblicazione biennale dal titolo Proteins sequence and structure.
a) Tecniche sperimentali.
L'approccio generale per determinare la sequenza degli amminoacidi di una proteina comprende vari stadi tra i quali: 1) preparazione del campione; 2) frammentazione della catena polipeptidica in piccoli peptidi; 3) separazione dei singoli peptidi e determinazione della loro sequenza; 4) allineamento dei peptidi in un'unica sequenza.
La purificazione della proteina comprende la separazione delle diverse subunità, l'apertura dei legami disolfuro e l'alchilazione dei residui di cisteina. Le subunità non identiche, tenute insieme esclusivamente da legami non covalenti (come per esempio nell'emoglobina), possono essere separate per precipitazione frazionata o mediante cromatografia a scambio ionico o gelfiltrazione in solventi che impediscono l'aggregazione. Queste tecniche possono essere applicate anche nel caso di subunità non identiche unite da ponti disolfuro (come nelle immunoglobuline) previa ossidazione o riduzione (v. eq. 1-3) e alchilazione dei residui cisteinici: l'apertura dei legami disolfuro tramite ossidazione con acido performico (v. eq. 2) trasforma sia la cistina sia la cisteina in acido cisteico (mentre l'apertura mediante riduzione con 2-mercaptoetanolo trasforma la cistina in cisteina). Tutte le cisteine inizialmente presenti e quelle prodotte per riduzione della cistina sono poi alchilate per evitare la loro ossidazione spontanea; gli agenti alchilanti più usati sono lo iodoacetato e la etilen-immina, che trasformano la cisteina rispettivamente in S-carbossimetilcisteina e S-amminoetilcisteina. Nell'ulteriore passaggio la catena polipeptidica viene spezzata in punti specifici, dando luogo a peptidi di cui si determina la sequenza; per spezzare la catena proteica si usano metodi chimici ed enzimatici. Tra gli svariati metodi chimici proposti, uno dei più soddisfacenti è quello che spezza la catena nei punti adiacenti ai residui di metionina per mezzo del bromuro di cianogeno: la reazione procede in modo praticamente quantitativo e poiché in genere vi sono pochi residui di metionina in un polipeptide, con questo metodo si ottiene una miscela di peptidi relativamente semplice. La reazione avviene nel modo seguente:
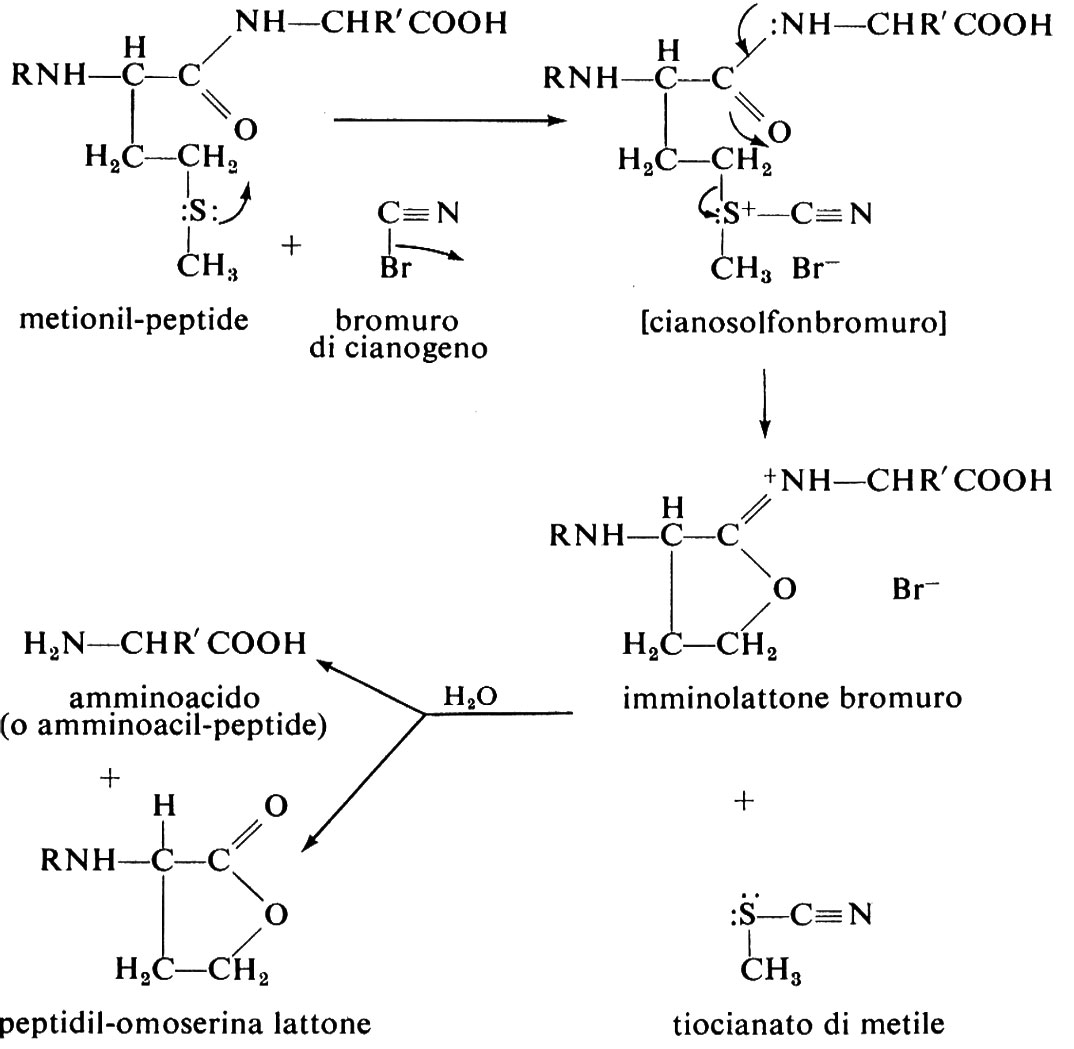
In teoria, da un polipeptide contenente N residui di metionina si ottengono N+1 peptidi, ciascuno dei quali contiene un residuo di omoserina, eccettuato il frammento carbossi-terminale. I peptidi ottenuti col bromuro di cianogeno vengono separati mediante precipitazione frazionata o per cromatografia e quindi frazionati ulteriormente, in genere attraverso un'idrolisi effettuata mediante enzimi proteolitici.
Gli enzimi proteolitici possono essere endopeptidasi (come la tripsina e la chimotripsina), che attaccano la catena polipeptidica all'interno, o esopeptidasi (come la leucinamminopeptidasi e la carbossipeptidasi), che staccano amminoacidi dalle estremità della catena. Le endopeptidasi tripsina, chimotripsina e termolisina sono relativamente selettive per alcuni residui di amminoacidi e quindi producono un numero relativamente piccolo di peptidi; endopeptidasi con minore specificità (come la pepsina) si usano generalmente solo per frazionare ulteriormente piccoli peptidi.
Il passaggio successivo comporta la separazione dei peptidi, ottenuti chimicamente o enzimaticamente, mediante l'uso della cromatografia a scambio ionico, della gelfiltrazione o dell'elettroforesi su carta. La sequenza degli amminoacidi nei peptidi purificati viene determinata dal distacco sequenziale di amminoacidi dall'estremità amminoterminale, ottenuto con il metodo di P. Edman e, quando occorra, dall'estremità carbossi-terminale mediante digestione con carbossipeptidasi (v. sopra). Il metodo di Edman consiste nel far reagire il peptide con isotiocianato di fenile: si forma un feniltiocarbammil-peptide; per successiva ciclizzazione e idrolisi del PTH-(feniltioidantoil-) amminoacido si libera l'amminoacido ammino-terminale:
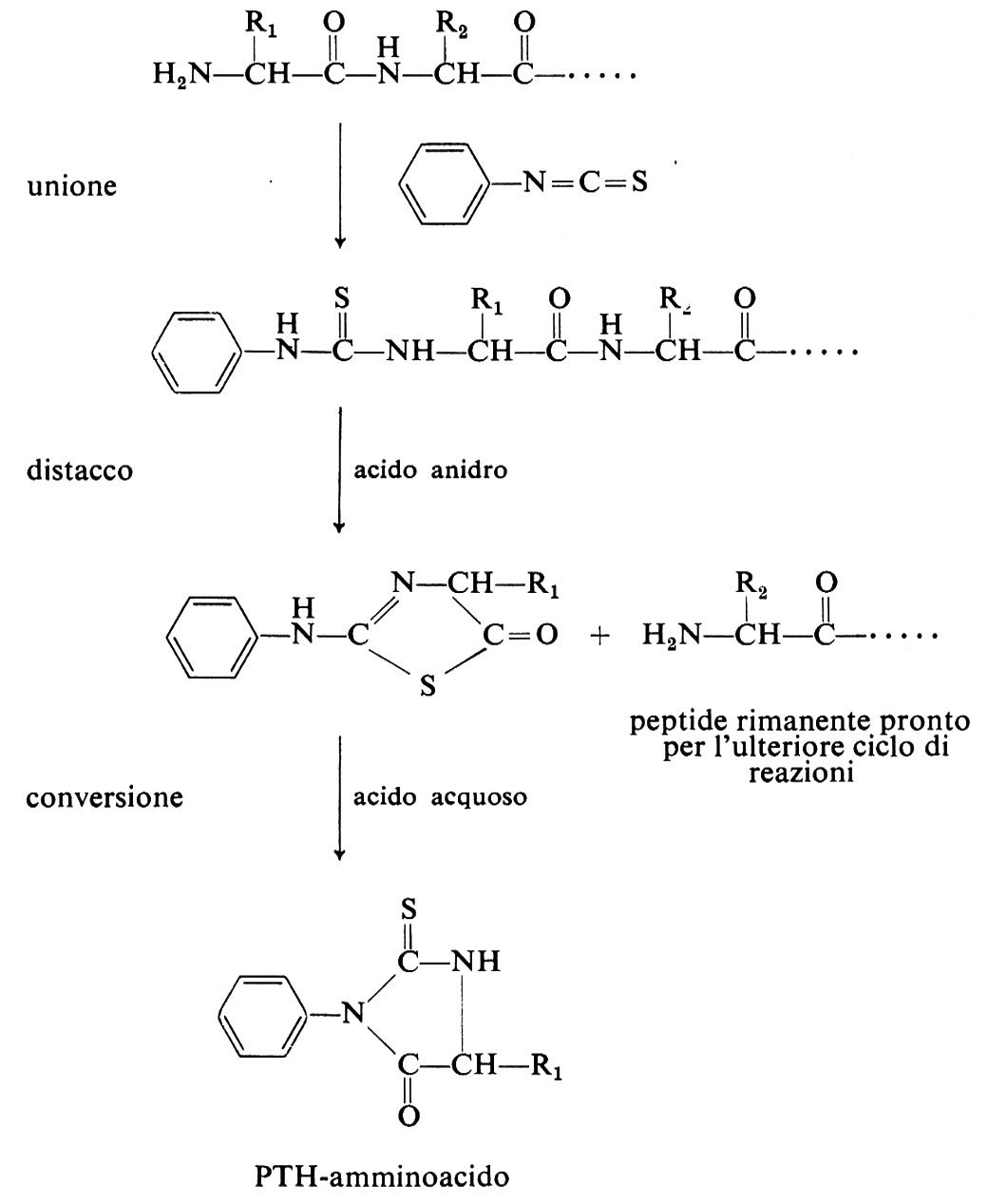
L'amminoacido ammino-terminale può essere identificato direttamente, individuando il PTH-amminoacido, o indirettamente, paragonando la composizione in amminoacidi del peptide prima e dopo ogni reazione con il reattivo di Edman. Di recente la degradazione di Edman è stata automatizzata.
Il passaggio successivo è l'allineamento dei peptidi ottenuti con due o più tecniche diverse col metodo della sovrapposizione, di cui si dà un esempio:
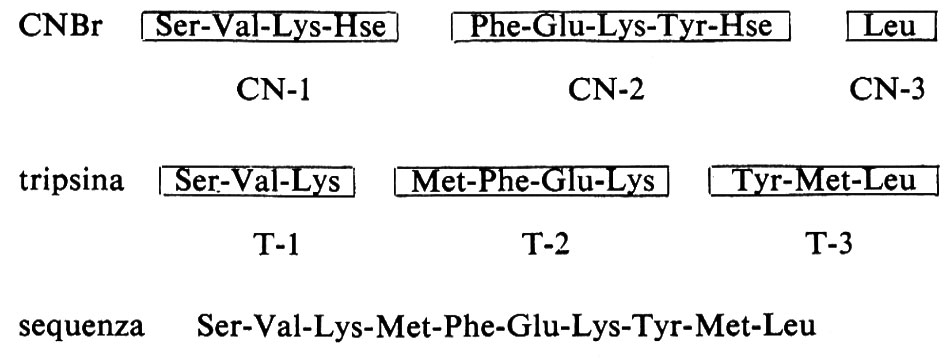
Il trattamento del decapeptide con bromuro di cianogeno produce i peptidi Ser-Val-Lys-Hse e Phe-Glu-Lys-Tyr-Hse e leucina libera, che dev'essere l'amminoacido carbossiterminale poiché da questa frazione manca l'omoserina (Hse); d'altra parte la digestione del decapeptide con tripsina produce i peptidi Ser-Val-Lys, Met-Phe-Glu-Lys e Tyr-Met-Leu; poiché la tripsina idrolizza i residui adiacenti alla lisina (e all'arginina), tutti i peptidi, a eccezione di quello carbossi-terminale, devono contenere lisina (o arginina) come residuo carbossi-terminale, il che è un corollario alla disposizione dei peptidi ottenuti con il bromuro di cianogeno sulla base dei residui di metionina; di conseguenza, il peptide carbossi-terminale deve essere Tyr-Met-Leu. Chiaramente le informazioni fornite da ciascuno dei due metodi non sono sufficienti da sole a determinare la sequenza dell'intero decapeptide. Invece i peptidi triptici possono essere disposti nell'ordine T-1-T-2-T-3, perché il frammento CN-2 ottenuto dal trattamento con bromuro di cianogeno si sovrappone al punto di separazione tra T-2 e T-3; nello stesso modo il frammento CN-1 si sovrappone alla regione compresa tra T-1 e T-2; è evidente quindi che nell'analisi sequenziale devono essere impiegati almeno due metodi diversi.
b) Verifica delle sequenze determinate chimicamente.
La sequenza degli amminoacidi della ribonucleasi è stata confermata mediante sintesi chimica della proteina enzimaticamente attiva usando sia i normali metodi chimici in soluzione (metodo in fase liquida) sia il metodo in fase solida; entrambi i metodi comportano la reazione di un amminoacido, il cui gruppo amminico è bloccato e il gruppo carbossilico attivato, con il gruppo α-amminico libero dell'amminoacido seguente, allungando così la catena a partire dal gruppo ammino-terminale. I gruppi reattivi delle catene laterali degli amminoacidi (come i gruppi ε-amminici della lisina, i gruppi carbossilici degli acidi aspartico e glutammico) devono essere protetti per impedire reazioni secondarie. Nel metodo tradizionale vengono sintetizzati peptidi di lunghezza intermedia, che poi vengono uniti tra di loro; il passaggio finale della sintesi consiste nel liberare tutti i gruppi bloccati. Come esempio di questo metodo viene illustrata, nella fig. 6, la sintesi del glutatione.
Nel metodo in fase solida, il gruppo carbossilico dell'amminoacido carbossi-terminale della catena peptidica viene legato a una resina; il vantaggio di questo metodo risiede nel fatto che eventuali impurezze e i reagenti in eccesso vengono eliminati per lavaggio a ogni passaggio, evitando così la necessità di purificare ogni peptide intermedio; il passaggio finale consiste nella rimozione di tutti i gruppi bloccanti e nel distacco del peptide dalla resina.
c) Informazioni ricavate dallo studio della sequenza.
Lo studio della sequenza degli amminoacidi non solo ha dimostrato che ciascuna proteina è un'entità a sé stante, ma ha anche fornito molte conoscenze sulle relazioni tra struttura e funzione e sul meccanismo di evoluzione delle proteine.
La sequenza degli amminoacidi in una proteina determina il modo in cui la catena polipeptidica è ripiegata nella sua conformazione biologicamente attiva: di conseguenza quando le proteine sono disciolte in soluzioni contenenti agenti denaturanti, come l'urea o i sali di guanidina, esse vengono denaturate, ma riacquistano la loro conformazione originale (‛nativa') quando questi agenti siano allontanati. Per esempio la ribonucleasi A è formata da una singola catena polipeptidica di 124 amminoacidi contenente quattro ponti disolfuro; in soluzioni contenenti urea e 2-mercaptoetanolo i ponti disolfuro vengono ridotti, l'attività enzimatica è completamente perduta e la proteina si comporta come un polipeptide di forma non organizzata; la rimozione dell'urea e la riossidazione graduale dei gruppi solfidrilici a disolfuro ripristina per il 95% l'attività enzimatica originaria: questo processo è illustrato nella fig. 7. Se i residui di cisteina della ribonucleasi ridotta riformassero a caso i ponti disolfuro, meno dell'1% delle molecole potrebbero riacquistare la struttura cataliticamente attiva; invece le molecole di ribonucleasi si riadattano nella conformazione originaria, in cui a ogni residuo di cisteina è stato posto di fronte quello ‛giusto' di un'altra cisteina per la successiva ossidazione della coppia.
Mutazioni in quel particolare segmento di DNA che codifica la proteina ne modificano la sequenza amminoacidica (v. Dixon, 1966); se si produce una proteina dalle caratteristiche migliori, la mutazione costituisce un vantaggio selettivo e può essere permanentemente incorporata nelle generazioni successive. Mutazioni ‛neutre' sono quelle che non producono proteine più utili o meno utili per l'organismo rispetto alla proteina originaria: queste mutazioni possono essere incorporate nel genoma tramite un processo detto ‛deriva genetica' (genetic drift). Alternativamente, se la mutazione produce una proteina meno utile per l'organismo, essa viene in genere eliminata dal genoma. Se la proteina è mutata al punto che l'organismo non può sopravvivere, la mutazione è detta ‛letale'; mutazioni meno gravi conducono a condizioni fisiologiche anormali (v. tab. IV; v. Garrod, 1963).

Un classico esempio dell'effetto della sostituzione di un amminoacido sulla funzione è l'emoglobina S, una variante dell'emoglobina (proteina trasportatrice di ossigeno) che si trova nei pazienti affetti da anemia falciforme: mentre l'emoglobina umana è composta da due catene α e due catene β (v. sangue: Emoglobina, Anemie emolitiche), l'emoglobina S ha due catene α normali, ma due catene β anormali, in cui una valina rimpiazza l'acido glutammico in posizione 6; da ciò deriva che l'emoglobina S è notevolmente meno solubile della normale emoglobina A e in effetti cristallizza all'interno dei globuli rossi con alterazione della forma dei globuli rossi stessi, il che produce una minore velocità del flusso sanguigno e l'anemia.
Lo studio della sequenza degli amminoacidi in una famiglia di proteine simili permette di identificare quali siano i residui ‛critici'. Un residuo può essere critico perché: 1) è necessario per un corretto ripiegamento della catena polipeptidica; 2) partecipa direttamente nel meccanismo di azione; 3) è coinvolto nelle specifiche interazioni tra subunità o tra proteina e proteina.
Il citocromo c, una piccola emoproteina (104 residui) che svolge un ruolo essenziale nel trasporto di elettroni, è stato isolato in più di 30 specie; un paragone delle sequenze in citocromi isolati da specie estremamente diverse, come i lieviti e l'uomo, rivela che circa un terzo dei residui è assolutamente ‛invariabile'; tali residui sono quindi critici per la funzione di questa molecola: tra questi sono quelli compresi tra le posizioni 70 e 80 e i leganti dell'eme Cys-14, Cys-17 e His-18. Oltre alle regioni invariabili, gran parte delle sostituzioni trovate sono conservative, cioè un amminoacido viene sostituito da un altro con caratteristiche chimiche simili, mentre altri residui sono ‛critici' nel senso che la loro sostituzione produce una proteina non attiva (v. tavv. I e II).
Il terzo tipo di informazione presente nelle sequenze di amminoacidi nelle proteine è la storia dell'evoluzione: poiché le linee filogenetiche che conducono alla scimmia e all'uomo si sono separate tra loro più recentemente rispetto a quelle che conducono ai lieviti e all'uomo, il citocromo c umano è molto più simile a quello della scimmia che a quello dei lieviti: in effetti è possibile costruire, sulla base delle differenze riscontrate nelle sequenze dei citocromi c, un albero dell'evoluzione (v. fig. 8) simile a quelli costruiti in precedenza sulla base di grossolane caratteristiche morfologiche; questa somiglianza conferma l'ipotesi che evoluzione delle proteine ed evoluzione morfologica procedano in modo parallelo.
Benché la velocità di evoluzione di una proteina (v. fig. 9) sembri essere relativamente costante, proteine diverse si evolvono a diverse velocità. Nella fig. 9 sono messe a raffronto le velocità di evoluzione del citocromo c, delle globine e dei fibrinopeptidi. La velocità di evoluzione riflette presumibilmente la capacità della proteina di tollerare sostituzioni di amminoacidi; questa capacità è funzione delle dimensioni della proteina in quanto: 1) le mutazioni sono di gran lunga più frequenti sulla superficie della proteina che nel suo interno; 2) il rapporto tra superficie e volume diminuisce con l'aumentare delle dimensioni molecolari. Quindi, in genere, le piccole proteine dovrebbero evolversi più velocemente delle grandi; tuttavia l'esistenza di esigenze strutturali in regioni situate alla superficie della proteina potrebbe diminuire la velocità di mutazione rispetto a quanto ci si aspetta sulla base delle sole dimensioni. Per esempio i fibrinopeptidi, che sono staccati idroliticamente durante la conversione del fibrinogeno in fibrina nel processo di coagulazione del sangue e che, una volta staccati, non hanno funzione fisiologica, possono tollerare grandi variabilità nella loro sequenza. L'emoglobina, invece, presenta minori variazioni perché devono essere conservati i residui critici per il normale ripiegamento della catena polipeptidica, per legare l'eme e per l'interazione tra le subunità. Il citocromo c, oltre a doversi conformare correttamente, deve interagire con altre proteine della catena respiratoria (citocromossidasi, per esempio) con interessamento di una parte relativamente grande della proteina e quindi vi è una ancor maggiore limitazione nella variabilità degli amminoacidi sulla sua superficie.
Infine la similarità osservata nella sequenza degli amminoacidi di proteine che svolgono funzioni differenti convalida l'ipotesi che tali proteine si siano evolute a partire da una comune proteina ancestrale tramite un processo denominato ‛duplicazione genica'. La probabilità che due proteine abbiano casualmente sequenze simili è infinitamente piccola. La duplicazione completa di un intero gene potrebbe produrre due regioni di DNA che portano lo stampo della stessa catena polipeptidica; uno dei due geni potrebbe mutare e formare catene polipeptidiche con proprietà diverse, mentre l'altro gene potrebbe ancora formare la proteina originale.
Le seguenti famiglie di proteine esemplificano casi di probabile duplicazione genica: 1) le proteasi ‛a serina': tripsina, chimotripsina, trombina, elastasi; 2) le carbossipeptidasi A e B; 3) le subunità delle immunoglobuline (IgG); 4) la α-lattalbumina (un componente della lattosiosintetasi) e l'enzima lisozima che idrolizza i polisaccaridi delle pareti cellulari; 5) la mioglobina e le catene α, β, γ e δ dell'emoglobina. Come esempio della duplicazione genica nella fig. 10 è rappresentato un meccanismo il quale spiega sia l'origine che l'evoluzione delle catene di globina.
Per riassumere, ogni proteina ha una sua tipica sequenza di amminoacidi; metodi per determinare queste sequenze sono stati approntati e usati in vari casi. La sequenza di una proteina è un'informazione basilare in quanto: 1) ne specifica la conformazione tridimensionale (v. molecole: Analisi conformazionale delle grandi molecole); 2) indica quali sono gli amminoacidi invariabili o critici; 3) dà notizie sullo sviluppo evoluzionistico della proteina.
6. Fattori che influenzano la conformazione proteica.
Una proteina, sciolta in soluzioni saline diluite, che mostri attività biologica e' considerata essere in forma ‛nativa'; in tali condizioni la conformazione di una proteina purificata è presumibilmente identica a quella della proteina allo stato naturale; tuttavia potrebbe non essere sempre così l'attività biologica richiede una conformazione specifica che, una volta modificata, per es. dalla presenza di agenti denaturanti o da temperature o pH non fisiologici, rende inattiva la proteina stessa. Poiché la conformazione originaria ha la maggiore stabilità termodinamica, cioè la minima energia libera, le conseguenze strutturali e funzionali dei cambiamenti di conformazione (cioè la denaturazione) sono spesso reversibili, come già detto.
Le interazioni delle varie catene laterali degli amminoacidi e dello scheletro polipeptidico tra di loro e con il solvente contribuiscono all'energia libera totale di ciascuna conformazione proteica. Il solvente può contenere piccole molecole di soluti (leganti) che si legano specificamente alla proteina stabilizzandone, a diversi livelli, le varie conformazioni: per esempio la carbossipeptidasi A può mostrare differenti conformazioni, con differenti proprietà funzionali, in presenza e in assenza di leganti.
Le interazioni che diminuiscono l'energia libera di una proteina possono essere classificate come apolari o polari. Le prime, dette anche idrofobiche, come accennato in precedenza, diminuiscono l'energia libera principalmente diminuendo l'entropia: il trasferimento di piccole molecole idrofobiche, simili a catene laterali di amminoacidi, da solventi apolari, come il benzene o il tetracloruro di carbonio, all'acqua consente di calcolare l'energia libera dei legami apolari. Questa reazione può essere considerata l'inverso del trasferimento di una catena laterale di un amminoacido idrofobico dalla soluzione libera all'interno di una molecola proteica. Sulla base di questi sistemi modello, si è visto che la formazione di legami apolari comporta un cambio di entalpia positivo, sfavorevole, che è però più che bilanciato da un notevole aumento di entropia. Questo aumento di entropia deriva presumibilmente da uno sconvolgimento della struttura dell'acqua, che si verifica quando il clatrato costituitosi intorno al residuo apolare viene distrutto e il residuo stesso viene trasferito in un ambiente non polare nell'interno della molecola. Il legame apolare è generalmente considerato il principale fattore termodinamico responsabile della struttura delle proteine. Gli amminoacidi leucina, isoleucina, fenilalanina, prolina, valina e, in minor misura, tirosina, triptofano e metionina, sono in grado di formare legami idrofobici e generalmente sono posti all'interno della molecola proteica, dove si trova il nucleo idrofobico. Poiché il trasferimento di un gruppo metilenico da un solvente non polare all'acqua comporta un cambiamento di energia libera di circa +1 kcal/mole, la presenza di un amminoacido non polare sulla parte superficiale di una molecola è molto sfavorevole dal punto di vista termodinamico.
Un secondo tipo di interazione che contribuisce all'energia libera è il legame polare; questo può essere costituito da un legame a idrogeno o da un ponte salino tra gruppi aventi carica opposta:
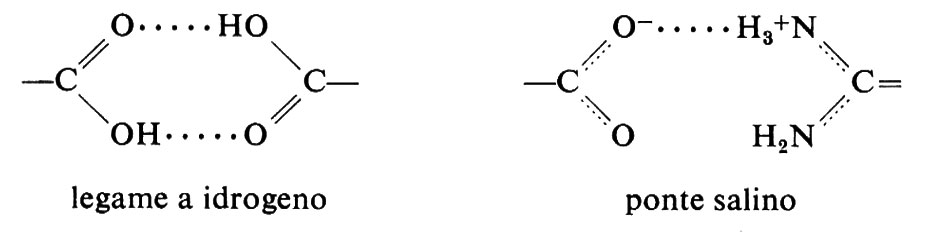
Questo tipo di legame era stato considerato di primaria importanza nel determinare il ripiegamento della catena polipeptidica, ma ora il suo ruolo è ritenuto secondario rispetto a quello dei legami idrofobici, in quanto i gruppi polari formano legami a idrogeno con l'acqua quasi altrettanto facilmente che tra di loro; essi probabilmente contribuiscono in modo significativo alla stabilità della struttura solo se si trovano racchiusi all'interno della molecola, dove non possono interagire con il solvente. Tale affermazione è suffragata dai seguenti fatti: 1) le molecole di N-metilacetammide si associano rapidamente tra di loro, per mezzo di legami a idrogeno, in solventi non polari, ma non in acqua; 2) strutture elicoidali stabilizzate da legami a idrogeno (come per es. il poli-γ-benzil-glutammato) si formano in solventi non polari, ma non in quelli polari; 3) analisi delle proteine per mezzo della diffrazione dei raggi X (v. sotto) mostrano che in genere i residui polari formano legami a idrogeno o ponti salini solo all'interno delle molecole. Ci si può immaginare una molecola di proteina come una gocciolina di grasso in un'emulsione: la molecola è composta da un nucleo formato principalmente dagli amminoacidi idrofobici, circondato dalle catene laterali degli amminoacidi polari che protrudono nel solvente.
La struttura proteica è influenzata anche dalla natura degli amminoacidi che compongono la molecola. La presenza di glicina e prolina è particolarmente notevole a questo riguardo: la prolina è un imminoacido e la sua presenza impedisce la formazione di alcune strutture elicoidali; la glicina, dal canto suo, non ha altra catena lateterale sul carbonio α che un atomo di idrogeno e quindi consente una completa libertà di rotazione, mentre tutti gli amminoacidi aventi catene laterali sul carbonio α limitano in modo notevole la rotazione stessa. L'orientamento relativo di due amminoacidi consecutivi in un polipeptide può essere definito mediante i due angoli ϕ e ψ (v. fig. 11); in base a ciò l'intera conformazione di una catena polipeptidica può essere espressa come valori di ϕ e ψ per ciascuno degli amminoacidi componenti. G. N. Ramachandran ha delimitato, sulla base di modelli, coppie di valori di ϕ e ψ entro regioni ‛possibili' e ‛impossibili'; un esempio di grafico secondo Ramachandran è riportato nella fig. 12, in cui le strutture totalmente possibili corrispondono alle zone delimitate da linee continue, mentre quelle parzialmente possibili corrispondono alle zone delimitate da linee tratteggiate. Un esempio di struttura impossibile è ϕ=0° e ψ=180°, che produrrebbe un'inaccettabile sovrapposizione degli atomi carbonilici adiacenti. Nelle proteine la cui struttura è nota, gli angoli ϕ e ψ sono in zone possibili o parzialmente possibili.
7. Metodi per determinare la struttura tridimensionale.
Forse il principale risultato nella biochimica delle proteine è stato la determinazione della struttura tridimensionale di queste molecole. I tre principali metodi sperimentali per affrontare questo problema sono: 1) la cristallografia a raggi X; 2) la microscopia elettronica; 3) la chimico-fisica delle soluzioni. Poiché ognuno di questi metodi ha, come vedremo, pregi e difetti intrinseci, una comprensione completa della conformazione proteica richiede una combinazione di tutti e tre.
1. Cristallografia a raggi X. - Fin dal 1913 il metodo della diffrazione dei raggi X è stato impiegato nella determinazione della struttura di molecole relativamente piccole. Questo metodo è basato sul fatto che una luce, nel passare attraverso un cristallo, viene diffratta se la sua lunghezza d'onda è dello stesso ordine di grandezza delle distanze interatomiche. Si giunge alla conclusione che la diffrazione dei raggi X può essere considerata, come è illustrato nella fig. 13, una riflessione da parte di piani successivi nel cristallo. I raggi diffratti sono in fase, cioè vi è un massimo di diffrazione, quando la somma delle distanze AB e BC è un multiplo intero della lunghezza d'onda; ci sarà quindi un massimo in ogni punto in cui è soddisfatta l'equazione di Bragg:
nλ=2d sen ϑ.
Il solo metodo che permette la determinazione della struttura di molecole proteiche per mezzo della diffrazione dei raggi X è quello del cristallo rotante; poiché questo metodo richiede cristalli singoli relativamente grandi (circa 1 mm in ogni direzione), spesso occorre molto tempo per trovare le condizioni ottimali di crescita che forniscano cristalli adatti. I cristalli vengono montati in piccoli tubi capillari e fatti ruotare intorno a ogni loro asse sotto il fascio di raggi X; i massimi di diffrazione (v. fig. 14) vengono registrati con una macchina fotografica precessionale o analizzati automaticamente; poiché per molecole complesse come le proteine si devono analizzare molte migliaia di massimi di diffrazione, sono stati costruiti diffrattometri automatici collegati a calcolatori elettronici.
La posizione e l'intensità dei massimi di diffrazione forniscono informazioni che possono condurre alla determinazione della struttura nel modo seguente. Si può dimostrare che la densità elettronica ρ può essere espressa così:
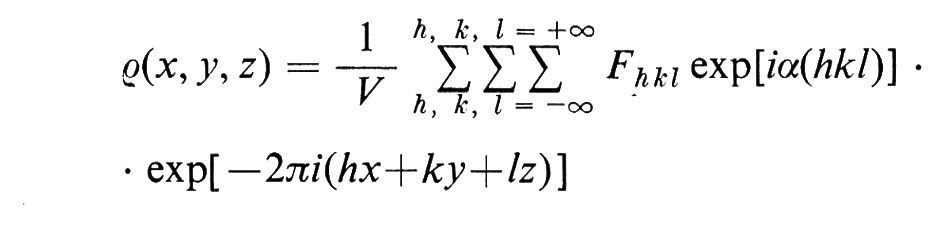
in cui V è il volume della cella elementare, h, k e l gli indici di Miller del piano responsabile della riflessione, determinato dalla posizione del massimo, e l'espressione Fhkl exp[iα(hkl)]•exp[−2πi(hx+ky+lz)] è in relazione alla intensità di ciascun massimo ed è funzione dell'intensità di diffusione da parte dei singoli atomi e dei loro rispettivi angoli di fase (α). Da questa espressione risulta evidente l'importanza di due fattori nel determinare i contorni di densità elettronica. Primo, più sono i termini (cioè i massimi) analizzati, migliore è il quadro di densità elettronica e quindi maggiore la risoluzione che se ne ottiene. Nello studio della mioglobina, con 400 termini analizzati si è ottenuta una risoluzione di 6 Å, ma sono stati necessari 9.600 termini per ottenere una risoluzione di 2 A; mentre la risoluzione di 6 Å fornisce un quadro generale dei ripiegamenti della catena polipeptidica, la risoluzione di 2 Å permette la localizzazione dei singoli atomi (eccetto l'idrogeno). Secondo, si deve conoscere l'angolo di fase α per computare i singoli termini della sommatoria; tuttavia l'intensità dei massimi è:
I=KFhkl•F*hkl
in cui
Feia•F*e-ia=FF*.
Quindi ogni riflessione ha un angolo di fase α non determinabile; cioè, in altri termini, l'analisi non ha un punto di riferimento. Questo problema nelle piccole molecole è superato per approssimazioni successive, assumendo le posizioni dei singoli atomi e calcolandone i valori di Fhkl che sono confrontati con quelli sperimentali. Tuttavia, poiché questo metodo non è applicabile in genere alle proteine a causa della loro complessità, si dovettero trovare nuove soluzioni al problema della fase: fra queste la più comunemente usata per fornire un punto di riferimento esatto che permetta di calcolare i singoli angoli di fase è quella della sostituzione isomorfa, che consiste nell'introdurre uno o più atomi pesanti, in ben determinate posizioni, in ogni cella elementare, senza alterare le dimensioni e la forma o la posizione degli atomi al suo interno. Poiché l'intensità della diffrazione da parte di un atomo è approssimativamente proporzionale al quadrato del numero di elettroni dell'atomo stesso, è possibile localizzare all'interno della cella elementare la posizione degli atomi pesanti; analizzando diversi derivati contenenti metalli pesanti è possibile calcolare gli angoli di fase e determinare la densità elettronica nella cella elementare.
Per mezzo della cristallografia a raggi X sono state determinate le strutture di numerose proteine, tra cui la mioglobina, l'emoglobina, la carbossipeptidasi A, la ribonucleasi e la chimotripsina, acquisendo così una migliore conoscenza delle relazioni tra struttura e funzione in queste proteine. L'organizzazione tridimensionale delle molecole proteiche è riportata nell'articolo successivo (v. proteine: Struttura tridimensionale).
2. Microscopia elettronica. - Benché la microscopia elettronica sia un importante metodo per lo studio della struttura e dell'organizzazione subcellulare, non è stata frequentemente applicata all'analisi della struttura proteica. I lati negativi di questa tecnica sono la risoluzione relativamente bassa (≈10 Å) e la necessità di osservare l'oggetto in condizioni drasticamente differenti dal suo ambiente naturale. Malgrado queste limitazioni, la microscopia elettronica è stata di grandissima utilità nell'ottenere modelli di molecole complesse, come l'actomiosina (la proteina contrattile del muscolo) e il collageno (la più importante proteina strutturale del tessuto connettivo). Questa tecnica ha anche mostrato la struttura di certi complessi enzimatici formati da molte subunità, come la piruvatodeidrogenasi, enzima responsabile dell'importante trasformazione del piruvato in acetil-coenzima A. L'enzima piruvatodeidrogenasi estratto da Escherichia coli ha un peso molecolare di 4,0•106 ed è composto da 88 catene polipeptidiche; nella fig. 15 sono riportate le fotografie al microscopio elettronico di questo complesso e la sua probabile struttura.
3. Chimico-fisica delle soluzioni. - Questo approccio sperimentale alla conoscenza della struttura delle proteine è basato sulle proprietà chimiche e fisiche delle proteine in soluzione: al contrario di quanto avviene nella cristallografia a raggi X e nella microscopia elettronica, la proteina viene studiata in uno stato fisiologicamente attivo. I metodi generali per determinare la struttura delle proteine in soluzione possono essere divisi in due categorie: 1) metodi che danno un'indicazione approssimativa della forma o conformazione della molecola; 2) metodi studiati per fornire informazioni riguardanti l'intorno e la reattività di specifici residui.
Le dimensioni e la forma generale di una molecola possono essere ricavate da proprietà idrodinamiche, come la viscosità, la velocità di sedimentazione, la diffusione. Misure di altro genere, come la dispersione ottica rotatoria, il dicroismo circolare e il dicroismo infrarosso o il magneto-dicroismo, permettono illazioni sulla struttura (contenuto in elica). Tra questi metodi sarà discusso come esempio quello della ‛velocità di sedimentazione'.
La velocità con cui una proteina sedimenta sotto l'influenza di una forza centrifuga dipende dal peso molecolare e dalla forma della molecola, dalla velocità angolare di centrifugazione e dalla composizione del solvente; di conseguenza si può prevedere che una molecola asimmetrica sedimenti più lentamente di una sferica, avente le stesse dimensioni, perché il suo volume di rivoluzione è maggiore. L'equazione di Svedberg stabilisce una correlazione tra velocità di flusso (u), velocità angolare (ω) e distanza dall'asse di rotazione (r) da una parte, e peso molecolare (M), volume specifico parziale del soluto (ã2), coefficiente d'attrito (f) e densità del solvente (ρ) dall'altra:
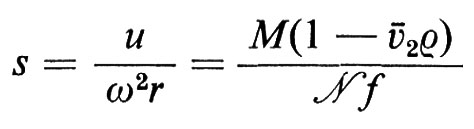
dove s, detta ‛costante di Svedberg', si determina sperimentalmente. Conoscendo M, ã2, e ρ si può calcolare f. Se la molecola fosse perfettamente sferica il coefficiente d'attrito (f0) sarebbe
f0=6πηR0
in cui
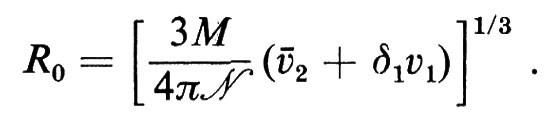
R0 è il raggio della sfera ed η il coefficiente di viscosità. Il termine δ1v1, che è la misura della solvatazione della molecola proteica, dev'essere incluso, perché il volume ‛attivo' della molecola comprende un alone esterno di idratazione. Il rapporto f/f0 può essere considerato una misura dell'asimmetria: valori intorno all'unità sono indicativi di molecole globulari compatte, mentre valori maggiori indicano lunghi filamenti o ellissoidi di rivoluzione.
La seconda categoria generale di metodi, comprendente la perturbazione del solvente, la titolazione degli idrogenioni, la risonanza magnetica nucleare e le modificazioni chimiche, fornisce informazioni circa i residui di amminoacidi. Per esempio, la modificazione chimica della mioglobina di balena in soluzione, ottenuta per reazione con bromoacetato, è stata utilizzata per distinguere i residui esposti al solvente da quelli racchiusi nell'interno della molecola: in condizioni specifiche di pH, il bromoacetato reagisce con velocità apprezzabile solo con i gruppi α-amminici e con le catene laterali di istidina e metionina; usando concentrazioni relativamente basse di reagente, la reazione è limitata ai residui superficiali della molecola proteica. L'analisi dei peptidi della mioglobina modificata evidenzia che il gruppo α-amminico e le istidine 12, 48, 64, 81, 113, 116 e 119 reagiscono con il bromoacetato e quindi presumibilmente sono esposte al solvente; invece le istidine 24, 36, 82, 93 e 97 e le due metionine non reagiscono e quindi verosimilmente sono all'interno della molecola. Un comportamento più o meno simile si manifesta quando il bromoacetato viene fatto reagire con la proteina cristallina: ciò fa ritenere che non vi siano grosse differenze di conformazione tra la mioglobina cristallina e quella in soluzione.
La cristallografia a raggi X, insieme alla chimico-fisica delle soluzioni, ha chiarito il meccanismo d'azione di numerosi enzimi (v. anche enzimi): ciò si è ottenuto esaminando comparativamente la diffrazione mostrata dalla proteina cristallina in presenza e in assenza di inibitori specifici. Questo metodo è indicato col nome di analisi differenziale di Fourier e mostra l'orientamento dell'inibitore nel sito attivo, cioè in quella parte dell'enzima che partecipa al legame e alla catalisi del substrato. La carbossipeptidasi A, per esempio, è un enzima la cui funzione specifica è di degradare sequenzialmente i peptidi a partire dall'estremità carbossilica; viene sintetizzata nel pancreas in forma inattiva (zimogeno) e viene attivata per idrolisi di uno specifico legame peptidico (v. enzimi). Unitamente alle altre peptidasi (enzimi che idrolizzano legami peptidici) presenti nell'intestino tenue, idrolizza le proteine alimentari ad amminoacidi. Il filamento polipeptidico della carbossipeptidasi A, determinato per mezzo della cristallografia a raggi X, è riportato nella fig. 16; la catena polipeptidica è costituita da 307 amminoacidi ed è ripiegata in modo tale da formare una molecola approssimativamente sferica di 52×44×40 Å; un atomo di zinco legato con i residui His-69, Glu-72 e His-196 è posto in una tasca a fondo cieco in cui si trova il sito attivo. Circa il 25% della catena polipeptidica è in forma di α-elica, presente in otto diversi segmenti (residui 14-29, 72-88, 94-103, 115-122, 174-184, 215-233, 254-262 e 288-305); approssimativamente un altro 20% della catena polipeptidica è costituito da otto segmenti disposti in struttura β (residui 32-37, 46-54, 61-67, 103-111, 190-197, 200-205, 238-243 e 265-271) sia parallela sia antiparallela, mentre la rimanente parte ha un orientamento irregolare e comprende un pezzo relativamente lungo a orientamento casuale (residui 122-174) e diversi segmenti più corti che uniscono i pezzi strutturati; un ponte disolfuro collega i residui 138 e 161 formando un'ansa verso l'esterno. Nella fig. 17 è riportato schematicamente il sito attivo della carbossipeptidasi A bloccato dall'inibitore glicil-L-tirosina; questo dipeptide è simile ai normali substrati e quindi, presumibilmente, si lega in niodo analogo, ma senza essere idrolizzato. Alcuni aspetti di questa struttura sono particolarmente interessanti. Primo, solo una piccola parte della molecola è direttamente coinvolta nella catalisi, mentre la restante parte serve a disporre i gruppi cataliticamente attivi nella corretta posizione. Secondo, la conformazione della carbossipeptidasi A è modificata notevolmente in presenza dell'inibitore: in particolare, la tirosina-248 ruota dall'esterno verso il sito attivo, orientando l'idrossile fenolico in vicinanza del legame peptidico da attaccare; questo movimento implica uno spostamento di circa 12 Å; anche il gruppo guanidinico dell'arginina-145 e quello γ-carbossilico dell'acido glutammico-270 si muovono di circa 2 Å; questi cambiamenti di conformazione sono i più rilevanti fra quelli osservati in seguito al legame fra un inibitore e un enzima. Infine, unitamente alla conoscenza della sequenza degli amminoacidi, l'analisi differenziale di Fourier ha permesso l'identificazione dei gruppi che partecipano alla catalisi; essi sono: lo zinco, l'acido glutammico-270, la tirosina-248 e l'arginina-145. Sulla base di queste informazioni è stato proposto un meccanismo d'azione secondo il quale l'α-carbossile libero del substrato si lega con un ponte salino all'arginina-145; lo zinco serve a polarizzare il carbonile del legame peptidico da attaccare, l'acido glutammico-270 agisce come base o nucleofilo e la tirosina-248 come donatore di protoni. Questo meccanismo è in accordo con le informazioni ottenute per altra via (modificazioni chimiche, cinetica) e sottolinea le possibilità della cristallografia a raggi X di svelare i particolari molecolari della catalisi enzimatica (v. catalisi enzimatica).
Benché a prima vista possa sembrare che la cristallografia a raggi X sia la metodica fondamentale per comprendere la struttura e la funzione delle proteine, in realtà non è così; le due maggiori limitazioni di questa tecnica sono: 1) che essa è statica; 2) che l'osservatore non puo sempre identificare le interazioni critiche. La prima limitazione risulta evidente nello studio della carbossipeptidasi A: benché la cristallografia a raggi X fornisca un eccellente quadro della molecola come tale e del suo complesso con l'inibitore, complesso verosimilmente analogo a quello fra enzima e substrato, non è possibile studiare i cambiamenti conformazionali che avvengono durante l'idrolisi enzimatica dei peptidi. Un esempio della seconda limitazione si trova nell'analisi strutturale del chimotripsinogeno e della chimotripsina. Il chimotripsinogeno, enzimaticamente inattivo, è convertito in chimotripsina attiva mediante rottura della catena polipeptidica tra i residui 15 (Arg) e 16 (Ile); essendo noti il sito attivo e il meccanismo d'azione, ci si aspetterebbe di veder chiarito il meccanismo di attivazione una volta confrontate le strutture del chimotripsinogeno e della chimotripsina; sfortunatamente le due molecole non presentano diversità, nelle regioni del sito attivo, che possano essere collegate alla funzione.
Una descrizione delle strutture dell'ossiemoglobina, della desossiemoglobina (emoglobina non ossigenata) e delle emoglobine patologiche fornisce un ottimo esempio delle capacità della cristallografia a raggi X. La funzione primaria dell'emoglobina, la principale proteina degli eritrociti, è il trasporto dell'ossigeno dai polmoni ai tessuti; un'importante funzione secondaria è la partecipazione al mantenimento di ragionevoli valori di pH nei tessuti: ciò è importante in quanto nei tessuti, durante l'ossidazione dei metaboliti, si accumulano quantità relativamente grandi di acidi (lattico e carbonico). Le seguenti proprietà rendono l'emoglobina singolarmente idonea al suo compito: 1) essa è molto solubile e ciò è indispensabile, in quanto ogni entrocita contiene circa 3•108 molecole di emoglobina. Come notato in precedenza, un'emoglobina anomala, l'emoglobina S, la cui solubilità è ridotta, produce un'alterazione nella forma degli eritrociti, responsabile dell'anemia falciforme (v. sangue: Anemie emolitiche); 2) la forma della curva di saturazione dell'emoglobina con l'ossigeno è sigmoidale e non iperbolica: quindi l'emoglobina lega facilmente l'ossigeno alle alte pressioni di ossigeno esistenti nei polmoni, ma lo cede facilmente alle pressioni minori riscontrabili nei tessuti; 3) la curva di saturazione con l'ossigeno è funzione del pH: l'ossigeno è ceduto più rapidamente ai valori più bassi di pH, che si hanno nei tessuti; questa proprietà è detta ‛effetto Bohr'. Poiché la forma desossigenata è una base più forte della forma ossigenata, la desossigenazione dell'emoglobina facilita pure l'allontanamento degli idrogenioni dai tessuti periferici; 4) l'emoglobina lega specificamente fosfati (come il 2,3-difosfoglicerato), il che provoca una diminuzione di affinità per l'ossigeno, facilitandone il distacco (v. sangue: Emoglobina). La molecola di emoglobina è un tetramero formato da due catene α e due β, ciascuna delle quali contiene approssimativamente 150 amminoacidi; non vi sono ponti disolfuro e quindi le quattro subunità polipeptidiche sono tenute insieme da legami non covalenti; in ogni subunità vi è un gruppo eme, anch'esso legato in modo non covalente. Nella forma ossi-, l'ossigeno diventa il sesto legame dell'atomo di ferro (Fe2+), mentre gli altri cinque sono i 4 azoti del gruppo eme e l'azoto imidazolico dell'istidina E7.
La diffrattometria a raggi X dei cristalli di emoglobina ha verificato le suddette caratteristiche. L'emoglobina è quasi sferica (65×55×50 Å), con le catene disposte come se fossero i vertici di un tetraedro regolare (v. fig. 18). L'organizzazione delle subunità è tale da favorire le interazioni tra subunità diverse (α−β) rispetto a quelle tra catene uguali (α−α o β−β); le interazioni dominanti tra subunità adiacenti sono quelle apolari piuttosto che quelle polari.
L'osservazione della conformazione delle singole catene mostra che le catene α e β sono molto simili tra loro e somigliano anche alla mioglobina, una proteina formata da una singola catena, che immagazzina l'ossigeno. Ciò non è sorprendente, in quanto le sequenze delle catene α e β dell'emoglobina sono molto simili a quella dell'unica catena della mioglobina.
Di particolare interesse è il grande contenuto di α-elica (75%) dell'emoglobina: ogni catena contiene diversi segmenti ad α-elica (indicati con le lettere da A ad H), uniti da brevi tratti non elicoidali (v. figg. 18 e 19). Tutti i segmenti elicoidali si approssimano al modello dell'α-elica con l'eccezione di C che è un'elica 310. I segmenti elicoidali si ripiegano a formare le singole subunità e questo ripiegamento forma in ogni subunità una tasca a forma di V, che serve a legare l'eme.
Infine è interessante notare la collocazione dei residui ‛critici' o invariabili. Solo sette residui invariabili si trovano in parecchie catene di globina: Gly-B6, Phe-CD 1, His-E7, Leu-F4, His-F8, Lys-H9 e Tyr-H22; la struttura tridimensionale dell'emoglobina ha parzialmente chiarito perché sono invariabili: Phe-CD 1, His-E7, Leu-F4 e His-F8 interagiscono in modo specifico con l'eme, mentre Gly-B6 e Tyr-H22 sono necessari per un corretto allineamento delle sezioni elicoidali; non è chiaro, invece, il ruolo apparentemente cruciale della Lys-H9.
Una completa comprensione delle conformazioni della desossi- e della ossiemoglobina ha consentito di delucidare la proprietà di ossigenarsi di questa proteina (v. Perutz, 1970; v. sangue: Emoglobina). Inoltre, le proprietà abnormi di numerose emoglobine anomale sono attualmente spiegabili facilmente col tipo di amminoacido sostituito e con la sua collocazione nella struttura tridimensionale: per esempio, la sostituzione, nell'emoglobina Torino, di una fenilalanina di una catena α (CD 1), che è implicata nel legare l'eme, rende la subunità α instabile, il che provoca clinicamente un'anemia. Quindi lo studio della struttura dell'emoglobina ha portato non solo alla comprensione della funzione di questa proteina estremamente interessante e importante, ma anche a una interpretazione, a livello molecolare, delle anormalità fisiologiche legate all'emoglobina (v. sangue).
8. Considerazioni conclusive.
Il nostro secolo ha visto spettacolari progressi nella comprensione della struttura e della funzione delle proteine. La vecchia convinzione che esse fossero macromolecole di composizione e struttura variabili e indefinite ha ceduto il passo alla convinzione che, come tutti gli altri composti biologici, esse abbiano proprietà chimiche e fisiche ben definite. La determinazione della sequenza degli amminoacidi di un ormone proteico (l'insulina) e di un enzima (la ribonucleasi) ha costituito il fondamento per la prova definitiva della struttura di queste e di altre proteine. L'analisi della sequenza ha anche delineato le vie dell'evoluzione molecolare e ha identificato gli elementi strutturali essenziali per la funzione biologica.
La diffrattometria a raggi X delle proteine ha ottenuto successi raramente eguagliati nella storia delle scienze biologiche. Negli ultimi tre decenni si sono superate difficoltà metodologiche apparentemente insormontabili ed è stata chiarita la struttura tridimensionale fino al livello atomico di molte proteine biologicamente importanti. Come risultato, noi ora comprendiamo a livello molecolare il meccanismo d'azione della mioglobina e dell'emoglobina nella respirazione, del lisozima, della chimotripsina, della carbossipeptidasi A e di numerosi altri enzimi nella catalisi enzimatica, di miosina, actina e tropomiosina nella contrazione muscolare, delle subunità enzimatiche interessate al controllo metabolico e delle proteine implicate nelle risposte immunitarie (anticorpi).
Mentre oggi sembra impossibile anticipare la struttura di una proteina dalle strutture già osservate, è possibile che una migliore comprensione dei concetti fondamentali della struttura, della funzione e dell'evoluzione delle catene polipeptidiche possa permettere di conoscere la conformazione conoscendo la sequenza degli amminoacidi (e viceversa). Prima di raggiungere questi obiettivi è necessario esaminare la sequenza di amminoacidi e la struttura tridimensionale di un numero di proteine molto maggiore di quello finora esaminato. A questo fine molto interesse è stato suscitato dall'introduzione di metodi automatici per l'analisi strutturale dei cristalli e per l'analisi della sequenza delle proteine.
Non è probabilmente una coincidenza che i maggiori contributi allo studio della struttura delle proteine siano stati portati da ricercatori con competenze diverse, variabili dalla biologia e dalla medicina alla chimica organica e dalla chimica fisica alla fisica; la complessità delle proteine è tale che solo uno sforzo comune, fatto utilizzando concetti e capacità sperimentali di vario tipo, avrebbe potuto condurre all'attuale livello di comprensione della struttura delle proteine e delle loro varie funzioni biologiche.
Bibliografia.
Dickerson, R. E., Geis, I., The structure and action of proteins, London-New York 1969 (tr. it.: Struttura e funzione delle proteine, Bologna 1973).
Dixon, G. H., Mechanisms of protein evolution, in Essays in biochemistry (a cura di P. N. Campbell, e G. D. Greville), vol. II, London 1966, pp. 147-204.
Fischer, E., Über einige Derivate des Glykocolls, Alanins und Leucins, in ‟Berichte der deutschen chemischen Gesellschaft", 1902, XXXV, pp. 1095-1106.
Garrod, A. E., Inborn errors of metabolism, Oxford 1963.
Hofmeister, F., Über Bau und Gruppierung der Eiweisskörper, in ‟Ergebnisse der Physiologie", 1902, I, pp. 759-802.
Pauling, L., Corey, R. B., Branson, H. R., The structure of proteins. Two hydrogenbonded helical configurations of the polypeptide chain, in ‟Proceedings of the National Academy of Sciences", 1951, XXXVII, pp. 205-211.
Perutz, M. F., Stereochemistry of cooperative effects in haemoglobin, in ‟Nature", 1970, CCXXVIII, pp. 726-739.
Perutz, M. F., Lehmann, H., Molecular pathology of human haemoglobin, in ‟Nature", 1968, CCXIX, pp. 902-909.
Sanger, F., Thompson, E. O. P., Kitae, R., Amide groups of insulin, in ‟Biochemical journal", 1955, LIX, pp. 509-518.
Svedberg, T., Pederson, K. O., The ultracentrifuge, Fair Lawn, N. J., 1940.
Watson, J. D., Molecular biology of the gene, New York-Amsterdam 1965, 19702 (tr. it.: Biologia molecolare del gene, Bologna 19722).
Struttura tridimensionale
SOMMARIO: 1. Alcune funzioni delle proteine. □ 2. Organizzazione strutturale. □ 3. Struttura secondaria. □ 4. Struttura terziaria. □ 5. Struttura quaternaria e aggregati proteici. □ 6. Gerarchia strutturale nelle proteine. □ 7. Modificazioni post-traduzionali delle proteine. □ Bibliografia.
1. Alcune funzioni delle proteine.
Le proteine sono i composti organici più rappresentati nelle cellule e nei tessuti animali. In un uomo di 70 kg, circa 12 kg sono proteine. Alla loro importanza quantitativa ne corrisponde una qualitativa ancora maggiore: quasi tutte le attività cellulari dipendono dalle proteine. Alcune hanno compiti esclusivamente strutturali, come il collageno e l'elastina dei tessuti di origine mesenchimale (connettivo, tendini, arterie) e le cheratine dell'ectoderma (capelli, peli, unghie), ma la maggior parte svolge funzioni complesse (v. tab. I). Le proteine enzimatiche sono catalizzatori specifici da cui dipende la velocità delle reazioni chimiche negli organismi viventi. Le proteine di trasporto legano selettivamente particolari composti per il loro immagazzinamento temporaneo o per il trasferimento da un tessuto all'altro. La trasformazione dell'energia chimica in lavoro biologico è affidata a proteine come la miosina e l'actina, che producono lavoro meccanico, come le pompe dell'Na+ e del Ca2+ che nelle membrane cellulari compiono lavoro osmotico trasportando ioni contro gradienti di concentrazione. Proteine partecipano ai sistemi di difesa dell'organismo; sono, infatti, di natura proteica gli anticorpi, il complemento e il fibrinogeno. Sono proteine alcuni ormoni, come l'insulina e le tropine ipofisarie, e anche alcune tossine batteriche, per esempio quella difterica. Le proteine hanno inoltre un'importanza primaria nell'alimentazione umana; da quelle di altri organismi derivano infatti alcuni amminoacidi che l'uomo non riesce a sintetizzare e che quindi devono essere introdotti con la dieta.

A tale molteplicità di funzioni deve necessariamente corrispondere una molteplicità di strutture. La diversità di forma delle macromolecole proteiche può già dare un'idea, sia pure grossolana, delle differenze esistenti tra proteine che compiono funzioni tra loro diverse. Per esempio, nelle cheratine e nel collageno, che hanno compiti esclusivamente meccanici, la disposizione spaziale (o conformazione) della catena polipeptidica è tale da formare delle lunghe fibre. La molecola del tropocollageno, l'unità proteica elementare delle fibrille del collageno, ha un peso molecolare di 300.000, è lunga 3.000 Å, ha un diametro di 15 Å ed è formata da tre catene polipeptidiche attorcigliate tra loro come i fili nelle corde. Al contrario, nelle proteine capaci di legare in modo specifico altre molecole, come gli enzimi e gli anticorpi, la catena polipeptidica si ripiega nello spazio e forma i ‛siti' di legame. Tali macromolecole hanno una struttura più compatta. L'emoglobina, che ha un peso molecolare di 64.500 ed è formata da quattro catene polipeptidiche, è pressoché sferica con le seguenti dimensioni: 65×55×50 Å. Per convenzione, indipendentemente dalle altre proprietà, si definiscono fibrose le proteine nelle quali il rapporto tra asse maggiore e asse minore è superiore a 10; le altre son dette globulari. In molte proteine sono presenti, associati alla catena polipeptidica con legami deboli o covalenti, ioni metallici o molecole organiche di varia natura, acidi nucleici, lipidi, glucidi, ecc. Abbiamo così le metallo-, le nucleo-, le lipo-, le glicoproteine. La porzione non proteica viene chiamata gruppo prostetico e le proteine che contengono gruppi prostetici sono dette proteine coniugate. Nuove proprietà nascono dall'unione della proteina con il gruppo prostetico.
Numerose proteine, semplici o coniugate, hanno la capacità di associarsi tra loro, e anche con altre macromolecole non proteiche, a formare strutture organizzate come i microtubuli, i complessi multienzimatici, i ribosomi, le fibrille, le membrane, ecc. che sono all'origine delle strutture subcellulari.
Come descritto nell'articolo precedente (v. proteine: Metodi di studio e struttura covalente), soltanto 20 amminoacidi concorrono, comunemente, alla formazione delle proteine. È dalla scelta e dalla ‛sapiente' distribuzione di tali amminoacidi nella catena polipeptidica che originano la struttura tridimensionale e le proprietà di queste macromolecole. La memoria cellulare, il DNA nucleare e mitocondriale, contiene il piano per la loro sintesi ribosomiale. Tale piano ha subito e subisce un'evoluzione continua di cui si trova traccia nella sequenza amminoacidica e nella struttura tridimensionale. Dall'evoluzione del piano, e quindi delle proteine, è nata e nasce la diversità strutturale e funzionale di queste.
2. Organizzazione strutturale.
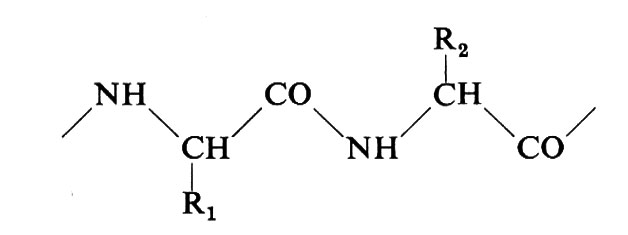
Secondo Linderstrøm-Lang e Schellman (v., 19592) l'organizzazione della molecola proteica può essere suddivisa in 4 livelli o strutture. La ‛struttura primaria' corrisponde alla struttura covalente della proteina: il numero delle catene polipeptidiche che la compongono, la loro sequenza in amminoacidi e la posizione dei ponti S-S. La catena polipeptidica può assumere nello spazio conformazioni che hanno carattere regolare (elicoidali o a pieghe) e che rappresentano la ‛struttura secondaria'. Il suo carattere periodico nasce dal periodico ripetersi nella catena di quella porzione degli α-amminoacidi che è comune a tutti (a esclusione della prolina e dell'idrossiprolina) e che dà origine allo scheletro peptidico:
La struttura secondaria è stabilizzata dai legami a idrogeno che con periodicità si formano tra i gruppi −CO− ed −NH− dello scheletro peptidico. La ‛struttura terziaria' compare nelle proteine globulari: la catena polipeptidica, che assume in vari tratti una struttura secondaria, si ripiega ulteriormente a formare una struttura tridimensionale nella quale residui amminoacidici lontani come sequenza vengono a trovarsi spazialmente vicini; è stabilizzata da legami deboli, a idrogeno, idrofobici, ionici, forze di van der Waals, che si formano prevalentemente tra le catene laterali dei residui amminoacidici. La ‛struttura quaternaria' si ha nelle proteine polimeriche, nelle quali più catene polipeptidiche (dette protomeri o monomeri o subunità) si aggregano tra loro mediante legami deboli analoghi a quelli che stabilizzano la struttura terziaria. Più recentemente, con il miglioramento della conoscenza delle proteine, sono stati inseriti tra la struttura secondaria e la terziaria altri due livelli strutturali: la ‛struttura supersecondaria', costituita da aggregazioni preferenziali di strutture secondarie, e il ‛dominio', porzione di catena polipeptidica che forma una ben separata regione globulare.
Vi sono infine i ‛complessi proteici' nei quali proteine, eguali o diverse, ciascuna con la propria struttura primaria, secondaria, terziaria, ed eventualmente quaternaria, ciascuna con una propria individualità funzionale, confluiscono per formare architetture più complesse con le quali si entra nell'ultrastruttura cellulare.
Dal punto di vista funzionale si può notare che con la struttura e la superstruttura secondaria compaiono le proprietà meccaniche; con il dominio e la struttura terziaria la capacità di riconoscimento specifico di altre molecole, la catalisi ecc.; con la struttura quaternaria le regolazioni più fini dell'attività proteica, la capacità di formare strutture tubulari ecc.; con i complessi le funzioni dei ribosomi, del citoscheletro, delle membrane, ecc.
3. Struttura secondaria.
La struttura primaria, le proprietà del legame peptidico, i metodi di studio delle strutture proteiche e i legami che le stabilizzano sono descritti in proteine: Metodi di studio e struttura covalente, al quale si rimanda il lettore.
Nelle proteine fibrose gli angoli ϕ e ψ (v. figg. 11 e 12 dell'articolo precedente) di numerosi residui amminoacidici assumono gli stessi valori, cosicché la catena polipeptidica si distende in una direzione, con andamento periodico. Le proteine fibrose rappresentano quindi dei modelli ideali per lo studio della struttura secondaria, che peraltro è presente anche nelle proteine globulari. Le più studiate sono le α-cheratine, in cui la catena polipeptidica è in α-elica destrorsa, il tropocollageno, che ha una struttura elicoidale sinistrorsa particolare - detta appunto ‛elica del collageno' - e la fibroina della seta, in cui le catene polipeptidiche hanno struttura β o a foglio pieghettato.
L'α-elica, proposta da Pauling, Corey e Branson (v., 1951), è una struttura elicoidale destrorsa con 3,6 residui amminoacidici per spira. Ogni residuo determina un allungamento assiale della struttura di 1,5 Å, per cui il passo dell'elica è di 5,4 Å.
I valori degli angoli ϕ (132°) e ψ (123°) sono tali da rendere minimi gli impedimenti sterici nello scheletro peptidico, da favorire forze di van der Waals attrattive tra gli atomi dello scheletro che si trovano in vicinanza all'asse dell'elica, e da disporre i gruppi −CO− ed −NH− dello scheletro a distanza e con orientamento ottimali per la formazione di legami a idrogeno che hanno direzione pressoché parallela all'asse della struttura. Inoltre essi indirizzano le catene laterali verso l'esterno dell'elica con un orientamento che ne diminuisce i possibili impedimenti sterici (v. fig. 1). A tale struttura corrisponde un contenuto energetico minimo e quindi una stabilità maggiore di quella che si riscontra in altre strutture elicoidali destrorse, come le eliche 310 (ϕ=131°, ψ=154°) e π(ϕ=125°, ψ=120°), che hanno rispettivamente 3 e 4,3 residui amminoacidici per spira.
Le eliche possono essere anche sinistrorse; tuttavia nelle catene polipeptidiche formate da L-amminoacidi le eliche destrorse sono più stabili.
Sebbene sia lo scheletro peptidico a determinare la struttura secondaria, anche le catene laterali dei residui amminoacidici svolgono un ruolo importante nello stabilizzare le eliche, come hanno dimostrato gli studi con polipeptidi sintetici ottenuti polimerizzando un solo tipo di amminoacido proteico. La poli-L-isoleucina non forma α-elica perché la catena laterale dell'isoleucina è troppo ingombrante e crea un impedimento sterico. Il poli-L-glutammato a pH 7 non ha struttura elicoidale perché le catene laterali cariche negativamente si respingono tra loro, mentre in ambiente acido la scomparsa delle cariche consente la formazione dell'elica. Analogamente, la poli-L-lisina acquista struttura ad α-elica solo in ambiente alcalino, quando scompaiono le cariche positive sulle catene laterali. Tra gli amminoacidi i cui polimeri non formano α-elica a pH 7 vi sono, oltre a quelli citati, l'aspartato, l'arginina, la glicina, la serina e la treonina. In questi due ultimi, la presenza degli idrossili alcolici nella catena laterale determina interazioni sfavorevoli alla formazione dell'elica. La poliglicina può formare diversi tipi di strutture elicoidali di cui una particolarmente importante perché è simile a quella del collageno. Tali amminoacidi, tuttavia, inseriti nelle catene polipeptidiche naturali alternati ad altri, partecipano alla formazione dell'α-elica pur rendendola meno stabile. La prolina, invece, non essendo un α-amminoacido, ma un imminoacido, non si accomoda nell'α-elica e ne interrompe di struttura.
L'α-elica destrorsa, detta semplicemente α-elica, è la struttura elicoidale più diffusa nelle proteine. Oltre che nelle α-cheratine, si riscontra per lunghi tratti, da 350 a 1.500 Å, corrispondenti rispettivamente a circa 65 e 280 spire, in altre proteine fibrose come la tropomiosina, la meromiosina e la paramiosina. Per tratti più brevi - da 1 a 8 spire con una media di 3 spire, corrispondenti a circa 16 Å - è presente in moltissime proteine globulari, nelle quali raramente e per brevi tratti, di i o 2 spire, si osservano anche eliche 310.
Crick (v., 1953), sulla base di studi cristallografici, ha postulato l'esistenza nelle α-cheratine di superstrutture secondarie. Due o più catene polipeptidiche ad α-elica sono tra loro attorcigliate a formare una superelica sinistrorsa con un passo di 140 Å. La superelica è energeticamente favorita dalla presenza, nei punti di contatto tra le eliche, di catene laterali idrofobiche. Quando i gruppi −NH2 terminali delle catene polipeptidiche si trovano alla medesima estremità la superelica è parallela, in caso contrario antiparallela. Tale superstruttura, in genere parallela, è estesa a tutta la molecola nelle sopracitate proteine fibrose e per brevi tratti in talune proteine globulari.
Il tropocollageno è formato da tre catene polipeptidiche parallele spiralizzate a elica sinistrorsa, con passo di 8,6 Å e 3 residui amminoacidici per spira (v. fig. 2). L'allungamento assiale è di 2,9 Å per residuo; I valori di ϕ e ψ sono rispettivamente 103° e 326°. Gli scheletri polipeptidici delle tre catene sono tra loro strettamente attorcigliati in una superelica destrorsa stabilizzata da legami a idrogeno perpendicolari all'asse della superelica. Tali legami si formano tra i gruppi −CO− ed −NH− dello scheletro peptidico di differenti catene. La struttura del tropocollageno è consentita dall'elevata percentuale di residui di glicina, prolina e idrossiprolina, che costituiscono, rispettivamente, il 33%, l'11% e il 12% circa della macromolecola. L'idrossiprolina (Hyp) è prodotta dall'ossidazione enzimatica di residui di prolina successivamente alla sintesi ribosomiale della catena polipeptidica. Il 95% circa delle catene ha la formula (Gly-X-Y)m, nella quale le posizioni X contengono frequentemente residui di prolina e le posizioni Y residui di idrossiprolina. È stato dimostrato sperimentalmente che i polipeptidi artificiali poli-glicina, poli-L-prolina e poli-L-idrossiprolina formano eliche sinistrorse con passo superiore a 9 Å e con 3 residui per spira. La presenza di glicina ogni 3 residui è determinante per la compattezza e la stabilità della struttura; la sua catena laterale, costituita da un solo idrogeno, si viene a trovare infatti nella zona di contatto delle tre catene, consentendone l'avvicinamento a una distanza utile per la formazione di legami a idrogeno intercatene.
Le molecole di tropocollageno si uniscono tra loro con legami deboli a formare fibrille che vengono ulteriormente stabilizzate da legami covalenti di cui parleremo successivamente. Sebbene la struttura del tropocollageno non sia stata riscontrata per ora nelle proteine globulari, è possibile che una struttura simile sia presente in una subunità del primo componente del complemento umano. Nel Clq, mediante la microscopia elettronica, è stato evidenziato infatti un raggruppamento di 18 catene parallele organizzate in 6 fibre collageno-simili (v. Reid e Porter, 1976).
La terza struttura secondaria è la struttura β, o a foglio pieghettato, ipotizzata anche questa da Pauling e Corey (v. fig. 3). Lo scheletro peptidico è disteso con un andamento periodico a zig-zag in cui ciascun segmento è costituito dai gruppi −CO−NH−, mentre i carbonî α si trovano nei punti di piegatura con la catena laterale diretta pressoché perpendicolarmente alla direzione dello scheletro peptidico. La periodicità è di 6,8 Å con 2 residui amminoacidici, per cui l'allungamento assiale per residuo è di 3,4 Å. Quando i valori di ϕ e ψ sono di 40° e 315°, l'assetto dello scheletro è tale che si possono formare legami a idrogeno con direzione perpendicolare sia a quella dello scheletro peptidico sia a quella delle catene laterali. Tali legami si formano tra gruppi −CO− ed −NH− di tratti di catene polipeptidiche che si affiancano con andamento antiparallelo a costituire una struttura supersecondaria, detta β antiparallela. Con valori di ϕ e ψ di 56° e 296°, l'assetto è tale che si formano legami a idrogeno fra tratti di catene parallele: si ha così la superstruttura β parallela, che è meno stabile perché la direzione del legame a idrogeno non è perpendicolare alla direzione dello scheletro peptidico. Sono anche possibili superstrutture miste parallele-antiparallele. Più frequentemente le catene a struttura β, invece di essere completamente distese, sono leggermente torte con un avvitamento sinistrorso molto dolce. Tale torsione provoca un aumento del periodo e del numero di residui amminoacidici per periodo, che salgono rispettivamente a 7,6 Å e 2,3, ma non impedisce la formazione della superstruttura e ne aumenta la stabilità. Nelle proteine globulari le strutture fi torte sono molto diffuse. I tratti sono tuttavia brevi, con una media di 6 residui corrispondenti a 20 Å, e in genere con 2-6 catene affiancate.
Una tipica proteina fibrosa in cui la struttura β si realizza per ampi tratti è la fibroina della seta (v. fig. 4). I dati cristallografici delle fibre di seta dimostrano che le catene corrono affiancate con andamento antiparallelo, sono leggermente torte e distano tra loro 4,7 Å, formando legami a idrogeno di 2,8 Å di lunghezza. Diversi piani costituiti da catene affiancate si sovrappongono a formare la fibra. La distanza tra i piani adiacenti è alternativamente di 3,5 e 5,7 Å. Questo consegue dalla particolare struttura primaria della fibroina, in cui si ripete l'esapeptide Gly-Ser-Gly-Ala-Gly-Ala. Le catene laterali della glicina si vengono così a trovare tutte dallo stesso lato del foglio pieghettato, mentre quelle di alanina e serina, più ingombranti, dal lato opposto.
Si osserva una buona corrispondenza tra le proprietà meccaniche delle proteine fibrose e la disposizione spaziale della catena polipeptidica. Nei capelli e nei peli, o nella lana (v. fig. 5), l'elasticità nasce dalla presenza delle α-eliche che possono allungarsi come delle molle, mentre la flessibilità è dovuta alle deboli interazioni tra le catene polipeptidiche. Una trazione esercitata in ambiente caldo umido ne provoca un allungamento duraturo dovuto alla formazione di β-cheratina, che ha struttura β-parallela. I legami a idrogeno intracatena delle α-eliche si trasformano in legami a idrogeno intercatene tipici delle strutture β. La β-cheratina, che è molto meno elastica della α, tende a riassumere spontaneamente la struttura elicoidale originaria per la presenza di ponti S-S e di una sequenza amminoacidica che favorisce la formazione dell'α-elica. Una caratteristica delle cheratine è la presenza di cisteina e di ponti S-S, da cui dipendono parte delle proprietà meccaniche. La maggior rigidità e anelasticità delle unghie e delle corna, formate anche queste da cheratina, è dovuta al maggior numero di ponti S-S. L'impiego in cosmetica di agenti riducenti, come l'acido tioglicolico, consente di rompere i ponti S-S delle cheratine dei capelli per renderli più flessibili e conferire loro la forma desiderata; con la riossidazione chimica dei gruppi −SH a disolfuri, tale forma viene fissata rendendola ‛permanente'. Anche nei processi di infeltrimento naturale o industriale della lana e di altri peli animali sono coinvolti i ponti disolfuro. Le conoscenze attuali sulle proprietà dei gruppi solfidrilici e dei ponti disolfuro devono molto alle ricerche condotte a scopo industriale sulle cheratine delle lane.
Il collageno è molto meno elastico delle cheratine. Le catene formano infatti eliche più allungate e sono tra loro strettamente attorcigliate nella superelica. Inoltre, numerosi legami covalenti tra le catene laterali delle supereliche irrobustiscono le fibrille di collageno fino a conferire loro la stessa resistenza meccanica alla trazione di un filo d'acciaio dello stesso peso. L'anelasticità della seta dipende dalla presenza della struttura β antiparallela: la catena è completamente distesa e l'allungamento lungo la fibra richiede la deformazione degli angoli di legame. L'assenza di legami covalenti tra le varie catene polipeptidiche che sono unite tra loro da soli legami a idrogeno e dall'incastrarsi delle catene laterali l'una a fianco dell'altra, conferisce alla seta la sua caratteristica flessibilità.
4. Struttura terziaria.
Con la struttura terziaria compare una proprietà fondamentale di alcune classi di proteine globulari (enzimi, anticorpi, proteine trasportatrici e recettoriali), quella di legare specificamente altre molecole (coenzimi, substrati, antigeni, ormoni, mediatori chimici, ecc.). Tale proprietà deriva dalla presenza di siti con struttura complementare alla sostanza che si lega.
Lo studio della struttura terziaria ha rappresentato uno dei più importanti e difficili problemi della biochimica. La cristallografia a raggi X ha consentito di raggiungere il formidabile risultato di definire la struttura tridimensionale delle proteine globulari e di gettare le basi per capirne il funzionamento. M. F. Perutz, J. C. Kendrew, D. C. Phillips, R. E. Dickerson e D. Hodgkin con i loro collaboratori hanno stabilito rispettivamente la struttura tridimensionale di emoglobina, mioglobina, lisozima, citocromo c e insulina (v. figg. 6, 7 e 8). Tra le numerose altre proteine di cui è nota la struttura tridimensionale ricordiamo la ribonucleasi, la subtilisina, la carbossipeptidasi A, la chimotripsina. Ogni anno il numero di proteine di cui viene determinata la struttura tridimensionale aumenta. La cristallografia a raggi X ha posto in evidenza come in molti casi la catena polipeptidica si ripieghi nello spazio a formare zone globulari compatte, separate tra loro da tratti non globulari. Tali tratti di connessione, più facilmente accessibili agli enzimi proteolitici, possono essere talvolta idrolizzati senza alterare la struttura e la funzione delle zone globulari. Queste osservazioni hanno fatto nascere il concetto di ‛dominio' strutturale come struttura globulare più elementare. Una proteina globulare può essere formata da uno o più domini. Il dominio contiene in genere da 100 a 150 residui amminoacidici e ha un diametro di 25 Å circa. Nel dominio la catena polipeptidica, che spesso assume nei vari tratti struttura secondaria α o β, si ripiega più volte nello spazio, acquisendo una ben definita e caratteristica struttura tridimensionale. Da un punto di vista del tutto generale, l'andamento complessivo della catena è paragonabile a quello che può assumere una fune che, tenuta verticalmente a un'estremità, venga fatta adagiare su una superficie. La fune si ripiega su se stessa ma non si annoda e può essere ridistesa risollevandola per l'estremità. La validità di questo paragone è confermata dall'assenza di ‛nodi' nelle proteine studiate.
In base alla loro struttura secondaria i dominî sono stati raggruppati in 5 classi. Alla prima appartengono quelli in cui è presente la sola struttura α, come nella mioglobina e nelle catene dell'emoglobina. Quando il dominio è costituito principalmente da tratti a struttura β, come quello della tripsina e quelli delle immunoglobuline, appartiene alla seconda classe. La catena polipeptidica, cambiando più volte direzione, forma un fascio di tratti affiancati costituiti da 3 a 10 residui amminoacidici a struttura β parallela o antiparallela. Le strutture antiparallele, più stabili, prevalgono e frequenti sono i meandri β (v. fig. 9). Nei dominî della terza classe i tratti ad α-elica sono raggruppati in una porzione della catena polipeptidica, mentre nell'altra è presente la struttura β, come si osserva nella ribonucleasi e nel lisozima dell'uovo di gallina. Appartengono alla quarta classe i dominî in cui tratti a struttura β si alternano a tratti ad α-elica; frequente è il ripiegamento di Rossmann (v. fig. 9). Tra le proteine con dominî della quarta classe vi sono la subtilisina e la carbossipeptidasi. I dominî che, come quelli della ferredoxina e della fosfolipasi, sono privi sia di struttura α sia di struttura β, sono raggruppati nell'ultima classe. Molte proteine sono formate da dominî appartenenti a differenti classi, come la glutationereduttasi, che possiede un dominio della terza classe all'estremità carbossilica mentre gli altri due, al centro e all'estremità amminica, sono della quarta classe (v. fig. 10). Fino a ora non è stata individuata una correlazione tra classe strutturale e funzione, anche se i dominî che legano i nucleotidi e i coenzimi nucleotidici appartengono in genere alla quarta classe.
La suddivisione dei dominî in classi è molto importante per comprendere i meccanismi che stanno alla base del ripiegamento della catena polipeptidica e l'evoluzione della struttura tridimensionale delle proteine.
Le proteine globulari sono costituite da uno o più dominî. Quando il dominio è uno solo, le sue proprietà funzionali si identificano con quelle della proteina. Negli altri casi, ciascun dominio può legare specificamente differenti molecole, come avviene nella glutationereduttasi (v. figg. 10 e 11). Un dominio lega il FAD, un altro il NADP e mediante il terzo, detto interfaccia, le due subunità dell'enzima si uniscono tra loro. Il sito catalitico, che lega il glutatione, è invece formato da più dominî. Nelle immunoglobuline la fissazione del complemento avviene mediante il dominio C2, mentre il sito di combinazione dell'antigene è costituito da due dominî, il VL e il VH (v. immunologia e immunopatologia). Nuove proprietà possono dunque nascere nelle zone di interazione tra i dominî. Inoltre la presenza in una stessa catena polipeptidica di più dominî può conferire alla proteina più funzioni, quali quelle di legare l'antigene e il complemento.
5. Struttura quaternaria e aggregati proteici.
Le proteine extraepiteliali che si trovano nei liquidi di secrezione, come gli enzimi digestivi del succo gastrico e pancreatico e il lisozima delle lacrime, sono piccoli monomeri in cui la struttura terziaria è irrobustita da numerosi ponti S-S. Il destino delle molecole secrete è molto incerto e l'ambiente in cui lavorano è scarsamente controllato, per cui conviene avere molte piccole molecole tra loro indipendenti e difficili da denaturare. Le proteine plasmatiche sono in genere di alto peso molecolare, sopra 50.000 dalton, per impedire che si disperdano nello spazio extravascolare o filtrino attraverso i reni. Possono essere monomeriche o polimeriche, ma le prime sono avvantaggiate perché non vanno soggette a fenomeni di disaggregazione se, per esempio, la loro concentrazione plasmatica diminuisce. Inoltre in proteine che, come le immunoglobuline, sono formate da più subunità, ponti disolfuro ne impediscono la disaggregazione per evitare sia la dispersione delle subunità più leggere sia il loro scambio con altre molecole simili ma dotate di differente specificità. Le proteine intracellulari sono di solito polimeriche, comunemente formate da poche subunità (proteine oligomeriche) unite tra loro da soli legami deboli. La membrana cellulare è infatti impermeabile alle subunità e non c'è quindi il rischio che queste vengano perdute. In realtà la difficoltà di sintetizzare sui ribosomi lunghe catene polipeptidiche è alla base della polimerizzazione proteica. Peraltro l'aggregazione dei monomeri ha i seguenti vantaggi: riduce la pressione osmotica intracellulare, consente di legare meno acqua per un più basso rapporto superficie/volume e rende meno viscoso il liquido intracellulare.
Nelle proteine oligomeriche compaiono spesso fenomeni di cooperatività nel legare substrati ed effettori, e ciò consente il controllo fine e rapido di attività metaboliche (enzimi allosterici) e del trasporto di ossigeno (emoglobina). Inoltre, la combinazione in rapporti diversi di subunità tra loro differenti consente la formazione di isoenzimi, da cui dipende in parte la differenziazione metabolica dei vari tessuti (v. enzimi). Dalla polimerizzazione di numerose subunità si ottengono in altri casi strutture fibrose come il filamento di F-actina, che si ha dalla polimerizzazione della globulare G-actina, e le fibre cave dei microtubuli, che si formano dalla polimerizzazione della tubulina, anch'essa una proteina globulare.
Negli aggregati proteici, più proteine globulari o fibrose, ciascuna dotata di una propria individualità strutturale e funzionale, si associano in rapporti e con geometrie ben definite, come avviene nella piruvicodeidrogenasi descritta nell'articolo precedente e nella sintetasi dell'acido grasso, un complesso costituito da sei enzimi diversi disposti a esagono con al centro una settima proteina che li tiene uniti. Con il complesso multiproteico nascono delle vere e proprie macchine molecolari. I complessi multienzimatici sopracitati sono paragonabili a catene di montaggio, in cui gli enzimi con la loro azione concatenata trasformano un composto in un altro senza che gli intermedi vengano dispersi nella cellula. La trasformazione dell'energia chimica dell'ATP in energia meccanica è consentita nel muscolo dalla ben definita architettura delle fibre di miosina e di actina.
Quali sono le basi strutturali dell'interazione tra i dominî nelle subunità, tra le subunità nelle proteine polimeriche, e tra le proteine negli aggregati? Abbiamo detto che intervengono legami deboli a idrogeno, ionici, idrofobici e forze di van der Waals, ma deve anche esistere una buona complementarità tra le superfici che interagiscono (v. fig. 12). Dall'analisi cristallografica risulta che la maggior parte dei residui non polari si trova localizzata all'interno del dominio, mentre i gruppi ionizzabili sono all'esterno, a contatto con il solvente. Tuttavia zone relativamente ampie, ricche di residui non polari, possono esistere anche sulla superficie. Tali aree tendono a legarsi più o meno labilmente con quelle che si trovano su altri dominî, subunità o proteine. Nell'area sono presenti gruppi polari che possono formare legami a idrogeno e ionici. Nel contatto fra aree diverse le interazioni di van der Waals sono in generale piuttosto numerose, in media un centinaio, i legami a idrogeno sono frequenti, più rari quelli ionici (v. tab. II). Nel determinare la complementarità delle superfici, la distribuzione dei legami a idrogeno e ionici è molto importante, come pure importanti sono le repulsioni elettrostatiche che possono opporsi all'aggregazione. Valga come esempio l'emoglobina S (HbS), una proteina patologica che a bassa tensione di ossigeno polimerizza formando lunghe fibre cave che deformano il globulo rosso diminuendone la resistenza meccanica (v. sangue: Anemie emolitiche). La modificazione della struttura primaria dell'HbS consiste nella sostituzione, nella posizione 6 della catena β, di un residuo di glutammato, carico negativamente, con uno non polare di valina. Nella HbS desossigenata sono presenti due siti complementari al residuo di valina e agli atomi a esso circostanti. Tali coppie di siti e di raggruppamenti danno luogo alla polimerizzazione (v. fig. 13). Il fibrinogeno è una grossa proteina plasmatica (90×450 Å; p. m. 340.000) che non ha nessuna tendenza a polimerizzare; una specifica proteasi a serina, la trombina, stacca alcuni frammenti, i fibrinopeptidi A e B, all'estremità di due delle tre coppie di peptidi che formano la molecola, trasformando il fibrinogeno in fibrina. La rimozione di tali frammenti, che rappresentano soltanto il 3% del fibrinogeno ma sono ricchi di cariche negative, provoca una nuova distribuzione delle cariche sulla superficie della molecola, favorendone la polimerizzazione.
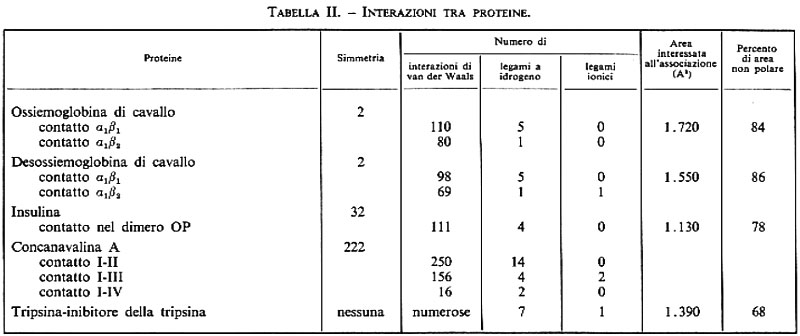
Dominî, subunità e proteine, associandosi, assumono spesso una disposizione simmetrica che ha destato grande interesse negli studiosi della struttura proteica. Gli stessi principî che si applicano alla descrizione di altre strutture simmetriche come molecole organiche, cristalli, ecc. sono utilizzati nelle proteine. Sebbene siano presenti simmetrie a gruppi spaziali - come nei cristalli dell'insulina dei granuli di secrezione delle cellule β delle isole di Langerhans nel pancreas e nelle proteine dei muscoli striati dei Vertebrati e degli Insetti - e simmetrie a gruppi lineari come nei microtubuli, nel virus del mosaico del tabacco e nei fagi filamentosi, la simmetria più frequente è quella a gruppi del punto. A causa dell'asimmetria degli amminoacidi non si osservano mai nei gruppi del punto centri di inversione o immagini speculari. Solo i gruppi n, n2, 23, 432 e 532 con n=1, 2, 3... sono possibili e, a eccezione del gruppo 23, esempi di tutti gli altri sono stati riscontrati nelle proteine (v. tab. III).
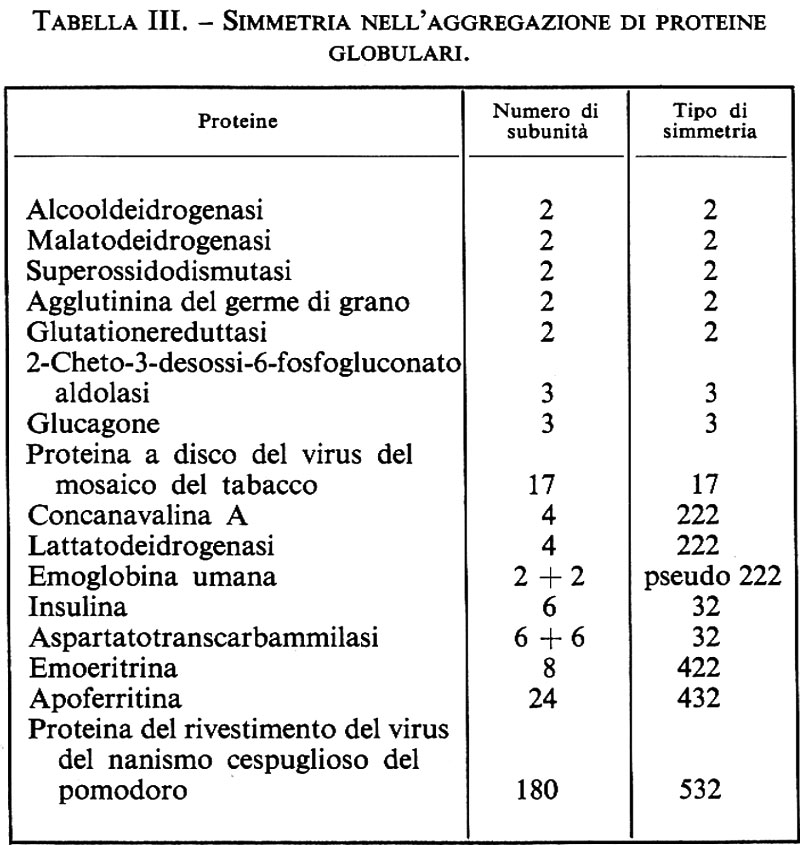
In alcuni casi si osservano aggregazioni pseudosimmetriche, in quanto i domin̄ o le subunità sono molto simili come struttura tridimensionale ma diversi chimicamente, come le subunità dell'emoglobina. Questa molecola tetramerica, formata da 2 subunità α e 2 subunità β, ha una vera simmetria 2 e una pseudosimmetria 222. Se le subunità fossero identiche avrebbe una vera simmetria 222. Se le subunità sono chimicamente identiche ma hanno conformazioni leggermente differenti si realizzano delle ‛quasi simmetrie' poiché i contatti tra le subunità sono leggermente diversi. La quasi simmetria è frequente nei virus sferici. Nel virus del nanismo cespuglioso del pomodoro, per esempio, le subunità sono formate da due dominî eguali, i quali tuttavia presentano una differenza conformazionale dovuta alla loro diversa disposizione spaziale: è così possibile ottenere un rivestimento icosaedrico che è quasi sferico, utilizzando un solo tipo di subunità.
La conoscenza della struttura tridimensionale delle proteme globulari consente di affrontare su basi molecolari i rapporti struttura-meccanismo d'azione-funzione biologica di queste macromolecole. Alcuni esempi di tali studi sono riportati dettagliatamente negli articoli enzimi; sangue: Emoglobina; tessuto muscolare; immunologia e immunopatologia, ai quali si rimanda il lettore desideroso di approfondire questo aspetto.
6. Gerarchia strutturale nelle proteine.
Gli esperimenti di denaturazione e rinaturazione dimostrano che la sequenza amminoacidica contiene le informazioni che consentono alla proteina di acquisire la sua struttura tridimensionale. Sebbene la suddivisione in livelli strutturali sia in parte artificiosa, ci si può chiedere se esista una gerarchia tale che ciascun livello determini quello successivo. Questa gerarchia è stata schematizzata nella fig. 14 in maniera estremamente semplificata. Si è infatti assunto che le interazioni avvengano esclusivamente tra i residui o tra gli elementi strutturali che si trovano vicini nella sequenza amminoacidica. Lo schema assomiglia a una piramide sociale nella quale i cittadini si organizzano in strutture sempre più complesse in base a un criterio strettamente territoriale. I cittadini che abitano vicini si organizzano in comuni, i comuni vicini in provincie, ecc. La qualità dei cittadini determina la qualità dei comuni, che a sua volta determina quella delle provincie e così via. In realtà, come in uno Stato moderno, nelle proteine, pur essendo spesso prevalente il criterio della ‛territorialità', vi sono interazioni anche tra amminoacidi lontani, per cui il modello proteico è molto più complesso di quello schematizzato nella figura.
Indubbiamente tale rapporto gerarchico è facilmente individuabile tra struttura primaria e secondaria, come hanno dimostrato gli studi già citati con i peptidi sintetici, e tra dominî, subunità e proteine rispettivamente nelle proteine globulari, nelle proteine polimeriche e negli aggregati proteici. I dominî, le subunità e le proteine, infatti, possono essere separati mantenendo spesso la propria struttura e la propria funzione. Inoltre il dominio, in genere, si forma prima che tutta la catena polipeptidica sia stata sintetizzata sul ribosoma. Più difficile è stabilire se esiste un rapporto gerarchico tra struttura secondaria e supersecondaria e tra queste e il dominio. Infatti, mentre le α-eliche hanno una loro intrinseca stabilità, un tratto di catena polipeptidica a struttura β è stabile solo se interagisce con altri tratti, eventualmente lontani nella sequenza amminoacidica. In questo caso l'acquisizione della struttura secondaria e supersecondaria potrebbe essere contemporanea. Molti dati indicano che alcune strutture supersecondarie sono intrinsecamente stabili ma altre certamente no.
Nonostante i limiti sopra esposti, al momento attuale si può affermare che la struttura proteica è in larga parte gerarchica. Questa conclusione è di particolare importanza per lo studio del ripiegamento della catena polipeptidica e dell'aggregazione, in quanto alcune tappe possono essere studiate indipendentemente. Inoltre dà più valore ai vari tentativi di prevedere la struttura tridimensionale sulla base della sequenza amminoacidica. Si è ancora molto lontani da risultati soddisfacenti, ma arrivare a una metodica che consenta di risalire alla struttura tridimensionale partendo dalla sequenza amminoacidica (e viceversa) sarebbe di grande interesse teorico e pratico. Basti pensare che si conosce la struttura primaria di centinaia di proteine, mentre sono relativamente poche le strutture tridimensionali note, e che molte proteine si denaturano con estrema facilità o presentano problemi di cristallizzazione così complessi da rendere per ora remota l'acquisizione della loro struttura tridimensionale mediante la cristallografia a raggi X.
Una forte limitazione a questo tipo di approccio è l'osservazione che sequenze amminoacidiche diverse danno strutture tridimensionali simili. Per esempio, la superossidodismutasi (un enzima che catalizza la trasformazione dell'acqua ossigenata in acqua) e le immunoglobuline presentano tali analogie nel ripiegamento della catena polipeptidica da suggerire l'ipotesi che si siano evolute da un precursore ancestrale comune (v. fig. 15). La funzione biologica e la sequenza amminoacidica si sono differenziate fino a perdere ogni analogia, mentre la struttura terziaria è stata in gran parte conservata (v. Richardson e altri, 1976). Questa osservazione, confermata in altre proteine, è di grande interesse perché dimostra che il meccanismo di ripiegamento della catena è sottoposto a una fortissima pressione selettiva. L'acquisizione di un nuovo meccanismo di ripiegamento della catena è così importante da essere gelosamente conservato.
Tra i numerosi risultati ottenuti dagli studi sul ripiegamento delle catene polipeptidiche, ricordiamo i seguenti. La rinaturazione in vitro è spesso più veloce della sintesi ribosomiale, per cui i domini si possono formare prima che la sintesi della catena polipeptidica sia completata. Se la proteina ha subito modificazioni enzimatiche successive alla sintesi ribosomiale (modificazioni post-traduzionali), di solito la denaturazione è irreversibile. Un esempio classico è quello dell'insulina. La proinsulina, costituita da una catena polipeptidica di 81 residui, rinatura spontaneamente formando ponti disolfuro nella posizione corretta alla quale è associata l'attività ormonale, mentre l'insulina (2 catene polipeptidiche per un totale di 51 residui) ne è incapace. Infatti con il distacco di un frammento peptidico di 30 residui al centro della catena proinsulinica, che si ha nella trasformazione enzimatica proinsulina→insulina, una parte delle informazioni necessarie per l'acquisizione della struttura tridimensionale viene irreversibilmente perduta. In alcune proteine il mantenimento della struttura nativa dipende dalla presenza di leganti specifici, ioni metallici o altri gruppi prostetici, effettori o substrati. Talvolta la proteina assume la sua conformazione funzionale solo quando si associa al gruppo prostetico. Questo può avvenire quando l'interazione tra la proteina e il gruppo prostetico è molto estesa, come si osserva in alcune nucleoproteine, lipoproteine e glicoproteine nelle quali la componente non proteica predomina.
Il ripiegamento della catena polipeptidica va visto come una transizione reversibile del sistema catena-solvente da uno stato di energia libera più alta, proprio della catena denaturata (che ha una struttura a caso), a uno di energia libera più bassa, proprio della conformazione nativa. Nel ripiegamento, come pure nell'aggregazione, si va verso strutture ordinate e quindi verso una diminuzione di entropia. Per esempio a 37 °C il termine TΔScatena per una proteina globulare di 150 residui amminoacidici è di parecchie centinaia di kcal/mole. Perché la catena polipeptidica si ripieghi acquisendo la conformazione nativa, è necessario che tale energia venga compensata e a ciò contribuisce una forte variazione dell'energia entalpica della catena (ΔHcatena), dovuta principalmente ai legami a idrogeno e alle interazioni di van der Waals, e una considerevole variazione di energia libera del solvente (ΔGsolvente), che per una soluzione acquosa salma è di almeno 100 kcal/mole. Il risultante ΔGtotale=ΔHcatena−TΔScatena+ΔGsolvente è molto piccolo rispetto agli altri termini: 10 kcal/mole circa. L'equilibrio tra la conformazione nativa e quella denaturata è dunque fortemente dipendente dalla temperatura, per cui poche proteine globulari mantengono la conformazione nativa a 60 °C e quelle che la conservano dopo la bollitura sono rarissime eccezioni in cui la stabilità è dovuta all'altissimo contenuto di legami idrofobici.
7. Modificazioni post-traduzionali delle proteine.
In questi ultimi anni, con la maggiore conoscenza delle strutture proteiche e dei meccanismi di sintesi ribosomiale, è divenuto sempre più evidente che una proteina, dopo che è stata sintetizzata nel ribosoma, e talora durante la stessa sintesi, può subire modificazioni covalenti che determinano nuove proprietà o ne fanno scomparire altre non più desiderate. Tali modificazioni si dividono in due grandi categorie. Le prime portano a una perdita selettiva di alcune porzioni della struttura primaria; tale perdita è sempre irreversibile ed è ottenuta da enzimi proteolitici specifici (v. Neurath e Walsh, 1976). Appartengono a questo tipo le trasformazioni di zimogeni inattivi in enzimi attivi (v. enzimi) spesso organizzate a cascata per ottenere l'amplificazione di un segnale, come si ha nella coagulazione del sangue e nell'attivazione del complemento; la formazione degli ormoni glucagone e insulina dai precursori inattivi proglucagone e proinsulina; la trasformazione del protropocollageno in tropocollageno.
Quasi sempre l'attività biologica per la quale la proteina è stata sintetizzata compare dopo la proteolisi. Qual è allora il significato del precursore? La presenza delle porzioni della catena polipeptidica che successivamente verranno staccate è nella maggior parte dei casi indispensabile per completare quell'insieme di informazioni chimiche che consentono alla macromolecola di acquisire una ben definita struttura tridimensionale o di aggregarsi. Questo è il caso della proinsulina, del chimotripsinogeno, delle catene del protropocollageno e di molti altri precursori di enzimi, ormoni, ecc. A tale esigenza se ne aggiungono spesso altre: nei processi digestivi, le proteasi devono venire attivate solo dopo che sono state secrete per impedire la proteolisi delle proteine cellulari; un sistema difensivo, come la coagulazione del sangue e l'attivazione del complemento, deve intervenire solo in caso di pericolo; la proteolisi parziale consente la progressiva e ordinata formazione del rivestimento proteico del virus T-4. L'attività biologica in tutti questi casi compare o perché il peptide rimosso mascherava il preesistente sito attivo, come nel pepsinogeno, o perché tale sito si costituisce in seguito a lievi modificazioni della struttura tridimensionale provocate dal distacco del peptide, come nel tripsinogeno.
Altre volte una capacità funzionale scompare con la proteolisi parziale: nel protripsinogeno un peptide idrofobico rappresenta un segnale per la secrezione della molecola. Nella tossina difterica il peptide ha un duplice scopo: mascherare l'attività enzimatica e consentirne la penetrazione cellulare; una proteasi della cellula invasa attiva l'enzima che, bloccando la sintesi proteica, ne provocherà la morte.
L'altro gruppo di modificazioni post-traduzionali enzimatiche interessa le catene laterali e i gruppi terminali. A differenza della proteolisi, la modificazione può essere reversibile, come negli enzimi interconvertibili che vengono attivati o inattivati mediante l'attacco covalente di particolari segnali chimici alla molecola proteica. Spesso la proteina modificata conserva la capacità di rinaturare. Tali modificazioni sono state così suddivise.
1. Modificazioni dell'−NH2 terminale. Ne sono esempi l'acetilazione di molte proteine muscolari, la formilazione dell'emoglobina di lampreda, la metilazione di proteine ribosomiali in Escherichia coli. In numerose proteine che operano negli spazi extracellulari - come immunoglobuline, ormoni, enzimi del veleno di serpenti - il glutammato o la glutammina terminali si ciclizzano ad acido pirrolidoncarbossilico. La modificazione può rendere l'estremità amminica meno polare o meno suscettibile alla proteolisi a opera delle amminopeptidasi. Nelle proteine polimeriche può rappresentare un mezzo per diminuire le interazioni tra le subunità, come si osserva nell'emoglobina di alcuni pesci.
2. Modificazione del −COOH terminale. La sola che si conosca è la trasformazione del carbossile in gruppo ammidico. Si osserva in alcuni ormoni, la secretina per esempio, e nelle proteine del veleno delle api. Anche in questo caso la funzione può essere o quella di proteggere l'estremità dalle carbossipeptidasi o di renderla meno polare.
3. Modificazione di catene laterali. L'idrossilazione di Pro e Lys nel collageno, la metilazione di His e Lys nella miosina, la carbossilazione di Glu nella protrombina, la fosforilazione di Ser e di Thr nella fosforilasi e nella caseina rappresentano solo alcuni esempi. Mediante tali modificazioni la proteina acquisisce nuovi residui amminoacidici, non codificati dal codice genetico, con la comparsa di nuove proprietà: la protrombina lega ioni Ca2+ solo dopo che è stato carbossilato il Glu; la fosforilazione determina l'aggregazione della fosforilasi e un forte aumento dell'attività enzimatica.
4. Formazione di legami covalenti con gruppi prostetici o altre molecole organiche; anche in questo caso possono comparire nuove proprietà spesso largamente dipendenti dal gruppo prostetico, come nel caso della biotina legata covalentemente a Lys nel sito attivo dell'enzima acetil-CoA-carbossilasi. Altre volte, invece, sono le proprietà della molecola proteica a venire modificate. Un esempio si ha nella glutamminasintetasi, nella quale l'adenililazione di un residuo di tirosina per subunità modifica alcuni parametri cinetici e di regolazione. L'ADP-ribosilazione del fattore EF2 della sintesi proteica, catalizzata dalla tossina difterica, causa il blocco della sintesi delle proteine. I radicali glucidici legati a Ser, Thr o Asn nelle glicoproteine svolgono un'ampia varietà di funzioni: impediscono la perdita di proteine plasmatiche attraverso il rene, abbassano il punto di congelamento nei pesci antartici, favoriscono e mantengono la distribuzione asimmetrica delle proteine di membrana, rappresentano dei sistemi di riconoscimento, ecc. Talvolta, come nelle mucoproteine in cui il gruppo prostetico polisaccaridico costituisce anche l'80% della struttura, la funzione della proteina diviene secondaria. In molti casi la distinzione tra un gruppo prostetico legato covalentemente o legato con legami deboli non ha una fondata giustificazione funzionale. La differenza più importante e significativa è questa: una molecola organica che venga legata covalentemente a opera di un enzima non ha bisogno di un sito di legame complementare e può svolgere la sua azione sulla superficie della proteina; una molecola associata con legami deboli deve avere invece un'ampia superficie complementare di contatto e spesso è contenuta in una tasca.
5. Formazione di legami covalenti tra catene laterali. I ponti S-S non vengono classificati tra le modificazioni post-traduzionali perché la loro formazione non è enzimatica. La costituzione di legami −CO−NH− tra gruppi ε-NH2 di residui di lisina e γ-CONH2 di residui di glutammina con eliminazione di NH3 si ha nella stabilizzazione del fragile coagulo di fibrina e nella coagulazione dello sperma di Roditori. I legami crociati del collageno si formano tra aldeidi prodotte dalla deamminazione delle catene laterali di lisina e residui di istidina o lisina. Nell'elastina, una proteina mesenchimale fibrosa simile al collageno ma particolarmente elastica, l'elasticità è dovuta a residui di desmosina che si ottiene dalla condensazione enzimatica di 4 residui di lisina, appartenenti a 2 catene polipeptidiche. La presenza di tali legami quadrupli consente alle fibre di riacquistare dopo lo stiramento la struttura originaria.
Il collageno offre un ampio e interessante panorama di modificazioni post-traduzionali (v. Tanzer, 1978). Nella formazione delle sue fibrille sono state individuate le tappe illustrate nella fig. 16. Le catene sono sintetizzate come procatene α1 e α2 in un rapporto 2:1. Ciascuna catena è formata da circa I .300 residui amminoacidici. Vengono idrossilati numerosi residui di prolina e alcuni di lisina. I residui di idrossilisina sono glicosilati con galattosio o con il disaccaride glucosio-galattosio. Nella formazione del trimero intervengono le estremità globulari delle tre catene che si aggregano e formano spontaneamente tra loro ponti S-S. Si ottiene così un perfetto allineamento delle porzioni fibrose, che possono spiralizzarsi formando la tripla elica. Successivamente, il protropocollageno viene secreto nello spazio intracellulare, dove le estremità globulari sono rimosse per proteolisi con la formazione di tropocollageno. Le molecole di tropocollageno si aggregano spontaneamente a formare le fibrille di collageno. Residui di lisina e idrossilisina sono deamminati ad aldeidi e formano legami crociati con altri residui di lisina e istidina.
Bibliografia.
Amzel, L. M., Poljak, R. J., Three-dimensional structure of immunoglobulins, in ‟Annual review of biochemistry", 1979, XLVIII, pp. 961-997.
Blake, C. C. F., Koenig, D. F., Mair, G. A., North, A. C. T., Phillips, D. C., Sarma, V. R., Structure of hen egg-white lysozyme, in ‟Nature", 1965, CCVI, pp. 757-763.
Blundell, T., Dodson, G., Hodgkin, D., Mercola, D., Insulin: the structure in the crystal and its reflection in chemistry and biology, in Advances in protein chemistry (a cura di M. L. Anson e J. T. Edsall), vol. XXVI, New York 1973, pp. 279-402.
Chothia, C., Conformation of twisted β-pleated sheets in proteins, in ‟Journal of molecular biology", 1973, LXXV, pp. 295-302.
Chothia, C., Janin, J., Principles of protein-protein recognition, in ‟Nature", 1975, CCLVI, pp. 705-708.
Crick, F. H. C., The packing of α-helices: simple coiled coils, in ‟Acta crystallographica", 1953, VI, pp. 689-697.
Frier, J. A., Perutz, M. F., Structure of human foetal deoxyhaemoglobin, in ‟Journal of molecular biology", 1977, CXII, pp. 97-112.
Kendrew, J. C., Dickerson, R. E., Stradberg, B. E., Hart, R. G., Davies, D. R., Phillips, D. C., Shore, V. C., Structure of myoglobin, in ‟Nature", 1970, CLXXXV, pp. 422-427.
Levitt, M., Chothia, C., Structural patterns in globular proteins, in ‟Nature", 1976, CCLXI, pp. 552-557.
Liljas, A., Rossmann, H. G., X-ray studies of protein interactions, in ‟Annual review of biochemistry", 1974, XLIII, pp. 475-507.
Linderstrøm-Lang, K. U., Schellman, J. A., Protein structure and enzyme activity, in The enzymes (a cura di P. D. Boyer), vol. I, New York 19592, pp. 443-510.
Neurath, H., Walsh, K. A., Role of proteolytic enzymes in biological regulation, in ‟Proceedings of the National Academy of Sciences", 1976, LXXIII, pp. 3825-3832.
Pauling, L., Corey, R. B., Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets, in ‟Proceedings of the National Academy of Sciences", 1951, XXXVII, pp. 729-740.
Pauling, L., Corey, R. B., Branson, H. R., The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain, in ‟Proceedings of the National Academy of Sciences", 1951, XXXVII, pp. 205-211.
Porcellati, G., Ricci, C., Ronca, G., Biochimica, Bologna 1978.
Reid, K. B. M., Porter, R. R., Subunit composition and structure of subcomponent Clq of the first component of human complement, in ‟Biochemical journal", 1976, CLV, pp. 19-23.
Richardson, J. S., Richardson, D. C., Thomas, K. A., Silverton, E. W., Davies, D. R., Similarity of three-dimensional structure between the immunoglobulin domain and the copper, zinc superoxide dismutase subunit, in ‟Journal of molecular biology", 1976, CII, pp. 221-235.
Schulz, G. E., Structural rules for globular proteins, in ‟Angewandte Chemie. International edition", 1977, XVI, pp. 23-33.
Takano, T., Kallai, O. B., Swanson, R., Dickerson, R. E., The structure of ferrocythocrome c at 2.45 Å resolution, in ‟Journal of biological chemistry", 1973, CCXLVIII, pp. 5234-5255.
Tanzer, M. L., The biological diversity of collagenous proteins, in ‟Trends in biochemical sciences", 1978, III, pp. 15-17.


