Tensioattivo
Enciclopedia on line
tensioattivo In chimica fisica, sostanza (detta anche sostanza tensioattiva) che, disciolta in quantità anche molto piccola in un liquido, fa diminuire notevolmente la tensione interfaciale che compete alla superficie di separazione fra la soluzione diluita così ottenuta e un’altra fase (solida, liquida o gassosa).
Meccanismo d’azione
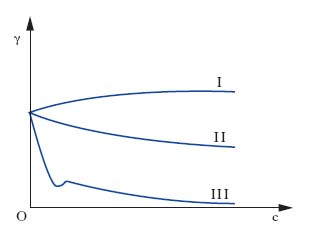
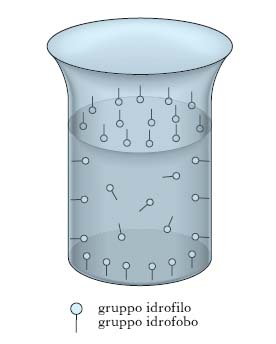
La fig. 1 illustra l’effetto che il progressivo aumento della concentrazione c di un soluto ha sulla tensione interfaciale γ di una soluzione: l’andamento I è presentato da sali inorganici (per es., NaCl), l’andamento II compete a composti organici solubili in acqua in tutti i rapporti (per es., glicerina), l’andamento III è tipico di composti organici in cui coesistono insieme gruppi liofobi e liofili (per es., oleato di sodio). L’andamento III, caratterizzato da una diminuzione assai notevole di tensione interfaciale all’aumentare di c, compete ai soluti t. e si giustifica con il fatto che essi presentano energie di interazione più basse rispetto a quelle che si manifestano fra le molecole di solvente: ciò provoca un accumulo del t. sulla interfase della soluzione in quanto le molecole di soluto, che per diffusione termica pervengono all’interfase, sono assoggettate a forze di attrazione verso l’interno del liquido meno intense di quelle che competono alle molecole di solvente e tendono, pertanto, a permanere sull’interfase. Questo comportamento dei soluti t. è spiegabile anche in relazione alla loro struttura; supponendo che il solvente sia acqua, come nella maggior parte dei casi pratici, le molecole dei t. risultano costituite da una parte polare (gruppo idrofilo, per es., un gruppo carbossilico, solfonico ecc.) che tende a sciogliersi in acqua con grande facilità e da una parte apolare (gruppo idrofobo, per es., una catena idrocarburica) che tende a rimanere insolubile. Pertanto le molecole dei t. mostrano la tendenza a raccogliersi sulle interfasi delle loro soluzioni, orientando verso l’interno il gruppo idrofilo (detto, pertanto, anche endofilo) e verso l’esterno il gruppo idrofobo (detto, pertanto, anche esofilo), come è evidenziato dalla fig. 2, che mostra come la concentrazione del t. disciolto in una soluzione raccolta in un Becher è maggiore non soltanto all’interfase liquido-gas, ma anche sulle pareti interne del Becher stesso (interfase liquido-solido).
Per mezzo di considerazioni termodinamiche J.W. Gibbs trovò (1876) la seguente relazione per la differenza Γ fra la concentrazione del soluto sulla interfase e quella nella massa interna della soluzione:
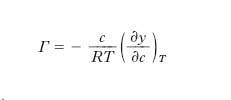
dove R è la costante dei gas perfetti, T la temperatura. L’isoterma di Gibbs (in cui, a rigore, le concentrazioni c dovrebbero essere sostituite dalle attività, ma l’errore che si commette è praticamente trascurabile nel caso di soluzioni diluite) rappresenta la conferma teorica del fatto che i t., a cui compete l’andamento III della fig. 1 caratterizzato da un valore negativo e molto elevato di (∂γ/∂c)T, hanno Γ positiva e molto grande, e si concentrano, pertanto, all’interfase, la cui tensione si abbassa fino a tendere a quella del soluto tensioattivo. Se la concentrazione del t. supera un determinato valore (concentrazione critica micellare), le molecole di t. non si sciolgono più nel solvente ma formano micelle colloidali, dotate di carica elettrica superficiale e conformate in modo che la parte esterna della micella risulti costituita dai gruppi idrofili delle molecole che si sono aggregate per formare la micella stessa, il cui interno, invece, è costituito dai gruppi idrofobi. Il valore della concentrazione critica micellare dipende dalla struttura molecolare del t. e in particolare dalla comparazione degli effetti del gruppo idrofilo e del gruppo idrofobo (che è lipofilo, cioè endofilo rispetto a sostanze non polari, come i lipidi): tale comparazione prende, pertanto, il nome di bilancio idrolipofilo ed è comunemente indicata con la sigla HLB (➔). Altri parametri che influiscono sul valore della concentrazione critica micellare sono la temperatura e la concentrazione degli elettroliti eventualmente presenti nella soluzione. La temperatura al di sotto della quale si ha la formazione delle micelle colloidali è detta temperatura di Krafft.
Funzioni principali
Sulla base delle proprietà indicate un t. può assolvere alle seguenti funzioni principali.
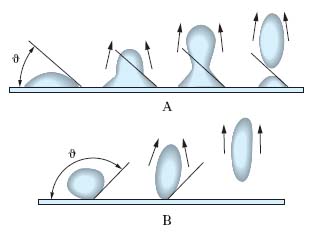
Detergenza. Si consideri un’impurezza liquida (per es., olio) su un supporto solido (per es., fibre tessili). Si immerga il sistema in un mezzo liquido. Se l’angolo di contatto ϑ delle gocce di olio sul supporto è <90°, l’effetto promosso da un’agitazione meccanica del mezzo liquido e tendente a staccare le gocce d’olio dà luogo a una pulitura soltanto parziale della superficie, sulla quale permangono goccioline, sia pur piccole, di olio (fig. 3A). Un t., aggiunto al mezzo liquido (L) diminuisce le tensioni interfaciali γSL con il solido (S) e γOL con l’olio (O), cosicché la relazione di equilibrio (➔ tensione) γSL=γSO+γOL cos ϑ, dove γSO è la tensione interfacciale fra il solido e l’olio, può essere soddisfatta soltanto per un valore di ϑ più elevato di quello corrispondente a un mezzo liquido senza t.; come risulta dalla fig. 3B, quanto più ϑ si avvicina a 180°, tanto migliore è la pulizia della superficie, cioè tanto più completo è il distacco delle impurezze oleose. Queste sono trattenute in sospensione nel mezzo detergente in quanto rimangono inglobate nell’interno delle micelle di t. insieme con le parti lipofile del t. stesso.
Quando il sudiciume depositato sul supporto è solido il problema si presenta assai più complesso in quanto la soluzione contenente il t. entra in contatto con due solidi di natura diversa, il supporto e il sudiciume, fra i quali si esercitano sia forze attrattive del tipo London-van der Waals sia forze repulsive elettrostatiche dovute al fatto che le superfici dei due solidi risultano caricate negativamente, con conseguente formazione di doppi strati elettrici caratterizzati da potenziali elettrocinetici di segno omonimo. I t. anionici (i più largamente impiegati) sono adsorbiti dalle superfici solide con cui entrano in contatto, aumentando, in valore assoluto, i corrispondenti potenziali elettrocinetici e conseguentemente accrescendo la repulsione elettrostatica fra i solidi fino a provocare il distacco del sudiciume; questo tende a portarsi verso il centro delle micelle di t., che possono così trattenere considerevoli quantità di impurezze solide. Sono state formulate anche altre teorie per spiegare il meccanismo della detergenza del sudiciume solido per mezzo di t., soprattutto nel caso dei t. non ionici (alcoli superiori etossilati, alchifenoli etossilati). Gli agenti t. si usano come detergenti (➔), sia per uso domestico sia per uso industriale (lavaggio di fibre tessili, bagni di tintura per favorire la penetrazione di coloranti ecc.).
Stabilizzazione. Con riferimento a sistemi costituiti da una fase A dispersa e da una fase B continua, quali aerosol (A: liquido; B: gas), emulsioni (A: liquido; B: liquido), schiume (A: gas; B: liquido), sospensioni (A: solido; B: liquido), i t., diminuendo la tensione interfaciale, aumentano la stabilità della dispersione di una fase nell’altra e, a parità di energia impegnata per l’agitazione del sistema bifasico, consentono di ottenere aree interfaciali maggiori. A seconda del tipo del sistema su cui agiscono, i t. assumono i nomi specifici di emulsionanti, flottanti, schiumogeni, bagnanti ecc. Agenti t. stabilizzanti trovano largo impiego nell’industria alimentare, dei cosmetici (per es., entrano a far parte della formulazione di shampoo), di prodotti farmaceutici, della carta, del cuoio, in agricoltura (assicurano la stabilità delle dispersioni di antiparassitari), nella flottazione dei minerali, nell’estinzione di incendi ecc.
Classificazione e scelte di utilizzo
I t. sono suddivisibili fondamentalmente in due categorie: t. naturali e t. sintetici. I t. naturali sono i saponi, mentre i sintetici provengono dalla petrolchimica e dagli oli vegetali. Alla fine degli anni 1990 si è andata affermando una nuova classificazione che prende spunto dalla precedente: la prima categoria di t. è costituita dai t. naturali e da quelli basati su fonti rinnovabili, come quelli che originano dagli oli vegetali, la seconda categoria è costituita dai t. che derivano dalla petrolchimica. A queste due categorie si deve aggiungere, in effetti, anche una terza categoria costituita dai t. ibridi, che derivano in parte dalla petrolchimica e in parte da t. naturali o da quelli ottenuti da fonti rinnovabili.
In relazione alle prime due categorie è osservabile un diverso atteggiamento da parte dei paesi industrializzati e da parte dei paesi in via di sviluppo. Nei paesi industrializzati, infatti, sino agli inizi degli anni 1990 si è fatto un utilizzo massiccio di t. di origine petrolchimica. Tale orientamento si è però successivamente modificato, con una sempre crescente tendenza a utilizzare t. quantomeno ibridi, se non addirittura di origine naturale o rinnovabile. Tale tendenza è ricollegabile alla migliore immagine che tali t. hanno nei confronti dell’opinione pubblica, dovuta alla loro maggiore tollerabilità, a un più elevato livello di biodegradabilità, a una minore pericolosità ambientale del processo produttivo, a un maggiore rispetto delle normative in tema ambientale. I paesi in via di sviluppo, a causa del loro ritardo tecnologico, sono ancora nella fase del passaggio dall’utilizzo dei t. naturali a quello dei t. di origine petrolchimica, che ottemperano meglio sia alle esigenze dell’industria sia a quelle dei consumatori. Solo quando questo primo processo di sostituzione sarà completato si potrà verificare quello attualmente in atto nei paesi industrializzati.
Lo scenario futuro del settore sembra destinato a mantenere questi caratteri almeno nel medio termine, sempre che l’industria dei t. di origine rinnovabile, segnatamente quelli di origine oleochimica, sia in grado di mantenere alto il livello di innovatività nei processi produttivi e di contenere il livello dei prezzi. Il quadro di lungo termine è invece ben più difficile da prefigurare, perché dipende dal grado di sviluppo dell’industria petrolchimica, strettamente legato alla disponibilità futura della materia prima petrolifera. In tal senso il processo evolutivo sopra delineato potrebbe necessariamente essere accelerato sia nei paesi in via di sviluppo sia in quelli industrializzati.
© Istituto della Enciclopedia Italiana - Riproduzione riservata


